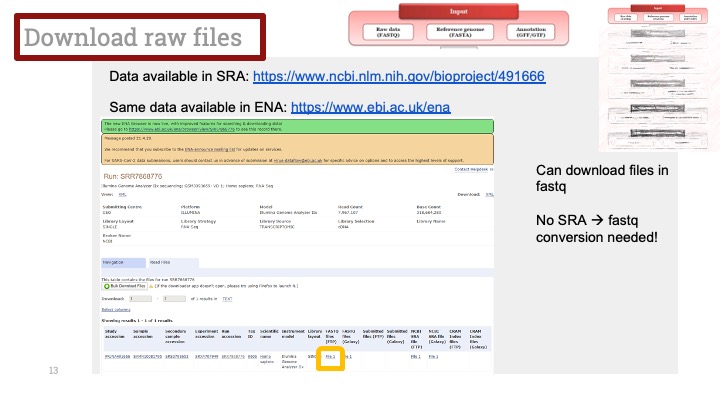
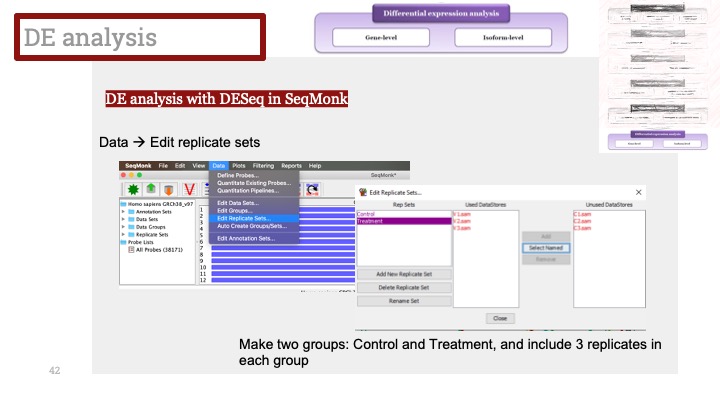
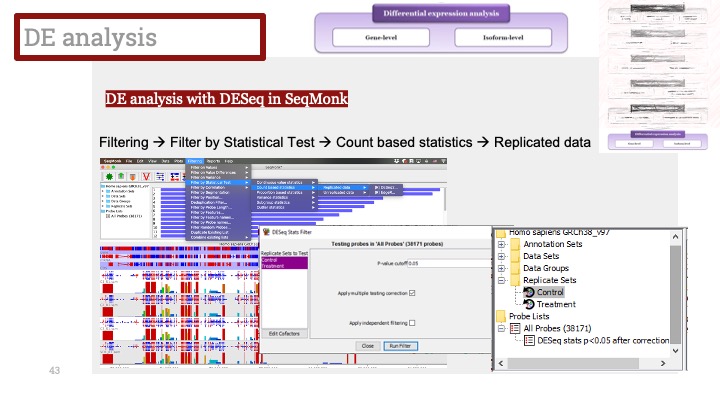
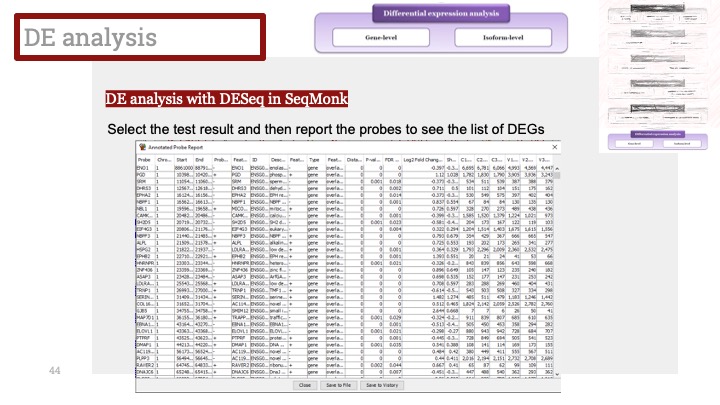
ALD는 Xq28에 위치한 ABCD1 유전자의 돌연변이로 인해 발생하며, X 염색체 열성 유전을 보입니다. ABCD1은 아주 긴 체인을 가진 지방산(very long chain fatty acid) 기질이 분해를 위해 퍼옥시좀(peroxisome)으로 이동하는 것을 도와주는 퍼옥시좀 막 운반체(peroxisomal membrane transporter)를 발현하는 유전자입니다. 이 유전자에 돌연변이가 생겨서 그 기능에 장애가 생기면 이 질병이 생기게 됩니다.
ABCD1 돌연변이를 가진 남성은 X 염색체가 하나이기 때문에 반접합성(hemizygous)을 지닙니다. 여성 보유자(female carrier)는 일반적으로 질병의 가장 심각한 증상은 보이지 않지만, 나이가 들어서 발병 증상을 보이게 됩니다. ABCD1 유전자의 돌연변이 발견 여부에 따라, ALD의 감염여부를 파악할 수는 있지만, 실제로 유전형과 표현형의 관련(genotype-phenotype correlation)은 없습니다. 한 가족 내에서, 동일한 유전자 돌연변이를 보유하더라도 각각 다른 형태의 질병 증상이 나타날 수 있습니다. 어떤 경우에는, 한 가족 내에서 감염된 환자 6명으로부터 5가지의 다른 질병형태가 나타난 예도 있습니다. ALD를 발병 시키는 공통적인 돌연변이는 없습니다. 대부분의 경우에 가족성 혹은 개인적인 특징을 띕니다. 거의 600가지에 달하는 돌연변이가 발견되었고, 그 중 절반 정도는 missesnse mutation이며, 1/4은 frameshift 돌연변이, 나머지는 in-frame deletion 혹은 splicing defect입니다. ALD에서 새로운 종류의 돌연변이가 나타나는 확률, 즉 부모로부터 물려받지 않고 새로이 나타나는 경우는 대략 4.1%이며, 이 경우 대부분은 생식계열 모자이크 현상(germline mosaicism)때문입니다.
발병기전
서로 다른 ALD 형태의 여러가지 증상에 대한 정확한 발병 원인은 확실하지 않습니다. 뇌의 백질(white matter), 고환의 라이디히 세포(Leydig cell), 부신피질(adrenal cortex) 등이 가장 영향을 많이 받습니다. 비록 그 증상은 국지적이더라도 과도한 양의 VLCFA(very long chain fatty acids)는 신체의 대부분의 조직에서 발견될 수 있습니다. Coenzyme A의 결핍은 VLCFA의 분해를 방해하고, 위의 조직 즉 뇌의 백질, 고환의 라이디히 세포, 부신피질 등에 그 축적을 초래하여 기관의 정상적인 기능을 막습니다. VLCFA의 농도는 로렌조오일(Lorenzo's oil)을 처방함으로써 정상화될 수 있지만, 질병의 진행을 막지는 못합니다. 그러나, VLCFA의 축적이 질병의 발병과 특정한 기작으로 관련이 있는지 혹은 생화학적 증상이 나타나는지 또는 질병의 식별과 관련이 있는지는 정확히 밝혀지지 않았습니다.
진단
ALD의 임상적 발현은 아주 다양하여서 그 진단이 어렵습니다. 다양한 형태로 인하여, 여러 종류의 증상을 종합하여 임상적 진단을 내리게 됩니다. ALD의 증상은 질병 형태 및 가족 구성원 사이 심지어는 쌍둥이 사이에서도 다양하게 나타납니다. 임상학적인 증상으로 인해 ALD가 의심되면 초기의 진단은 가스 크로마토그래피(gas chromatography)와 질량 분석(mass spectrometry)을 이용한 혈장 내 VLCFA(very long chain fatty acid)의 정도에 의해 결정됩니다. VLCFA 농도로 양성이 나온 후에 ALD의 확진은 ABCD1 유전자를 조사하여 내리게 됩니다. 여성의 경우에는 혈장 VLCFA 수치가 확정요소가 아니기 때문에 (어떤 여성 환자들은 VLCFA의 혈장 수치가 정상임) 특히 가족력이 있는 경우 유전자 진단이 선호됩니다. 남성은 임상적 증상이 아주 다양하지만, ABCD1 유전자 돌연변이를 가진 남성은 모두 VLCFA 수치의 증가를 보입니다.
ALD와 관련한 수치 증가는 임상적 증상이 나타나기 훨씬 전 출생 시 부터 나타나기 때문에, 신생아를 진단하는 다양한 방법들이 개발되어 있습니다. ALD와 관련한 신생아 진단의 어려움 중 하나는 환자가 어떠한 증상을 보일지 예측하기 힘든 것입니다. 유아기 대뇌 형태를 보이는 ALD에 감염된 남자아이들에게 널리 쓰이는 치료법은 골수 이식(bone marrow transplant)가 있지만, 이것은 또한 많은 위험을 동반하기도 합니다. 그러나 대부분의 남성 ALD 환자가 부신기능부전증(adrenal insufficiency)를 동반하기 때문에, 질병의 조기 발견과 그 치료는 증상의 심화를 막을 수 있고 이 후 다른 증상의 치료를 위한 관찰을 가능하게 합니다.
Loes score는 MRI로 보이는 대뇌 이상의 정도를 측정하는 등급입니다. 이는 0에서 34까지 있으며, 질병의 진행 정도와 증상 부위 및 뇌의 전반적인 혹은 국지적인 위축 정도에 의해 점수를 매기는 시스템입니다. 이 등급 제도는 신경방사선학 의사인 Daniel J. Loes에 의해 개발되었으며, 질병의 진행 정도와 치료의 효율성을 평가하는 중요한 척도가 됩니다.
치료
식이요법
ALD 치료에 대하여 초기의 식이요법은 VLCFA의 섭취를 제한하는 것이었습니다. 하지만, VLCFA는 체내에서 생성되기 때문에 식이적인 섭취가 유일한 근원이 아닙니다. 그러므로 VLCFA의 식이적 제한은 혈장 및 다른 체내 조직에서의 VLCFA 수치에 영향을 주진 않습니다. VLCFA의 체내 생성이 그 수치에 중요한 역할을 한다는 것이 밝혀지고 나서는, 식이요법은 이러한 생성 경로를 억제하는 데에 중점을 두게 되었습니다. ALD 환자였던 Lorenzo Odone의 부모는 질병의 진행을 늦추기 위한 식이요법을 개발하는 데에 엄청난 노력을 하였습니다. 그 결과, 그들은 로렌조 오일(Lorenzo's oil)로 알려진 불포화 지방산 혼합물(glycerol trioleate와 glyceryl treirucate 4:1의 비율)을 개발하였고, 이는 체내에서 포화지방산의 연장을 억제하는 것으로 알려져있습니다. 확실히 로렌조 오일의 섭취가 체내 VLCFA 수치를 낮춰주긴 하지만, ALD가 대뇌에 미치는 영향에 효과적인지 여부는 여전히 입증되지 않았습니다. 로렌조 오일의 처방은 이미 발병한 환자의 신경 감퇴 및 신장 기능 약화를 막지는 못하는 것으로 밝혀져 있습니다.
이식
식이요법이 ALD 환자에게서 VLCFA의 혈장 내 수치를 정상화시키는 데에는 효과가 있지만, 대뇌 ALD의 대표적인 증상인 탈수초화(demyelination)을 막는 유일한 치료법은 동종이계 조혈 줄기 세포(allogeneic hematopoietic stem cell)의 이식입니다. 이 방법이 효과적이기 위해, 이식 수술은 질병의 초기 단계에 행해져야 합니다. 만약 탈수초화(demyelination)이 진행되어버렸다면, 이식 수술이 그 진행을 악화시켜 탈수초화를 더 빠르게 진행시킬 가능성도 있습니다. 이식 수술이 ALD의 유아기 대뇌 형태 증상을 보이는 환자에게서 탈수초화 진행을 중지시키는 데 효과가 있지만, 이 환자들의 신장 기능을 개선시키지는 못하는 것으로 나타났습니다.
유전자 치료
이식 수술에 적절한 기증자가 나타나지 않는 환자들을 위하여, 유전자 치료를 이용한 방법 또한 연구 중입니다. ABCD1의 정상 형태를 발현하는 적절한 벡터를 선택하여서, 골수 이식 혹은 줄기 세포 이식과 유사한 방법으로 환자에게 이식합니다. 유전자 치료는 주로 프랑스에서 소수의 환자들에게만 시행되었습니다. 이 환자들은 기존의 이식 방법에 HLA match가 없다고 밝혀진 후에야 유전자 치료가 시행되었으며, 이중 두명의 환자에게서 탈수초화 증상이 2년까지 나타나지 않았다고 보고된 바 있습니다. 그러나, 유전자 치료가 신경학적인 증상의 치료에는 성공적이었지만, VLCFA 혈장 수치는 그대로 높아진 상태였습니다.
부신기능부전증
남성 ALD 환자에게서 자주 보이는 부신기능부전증(adrenal insufficiency)의 치료는 신경세포에서 나타나는 증상 완화와 관련이 없어보입니다. 부신기능부전증이 나타나는 환자에게는 호르몬 교체(hormone replacement)가 일반적입니다. 부신기능부전증은 이식 수술이 성공적이더라도 완화되지 않으며, 이로 인해 여전히 호르몬 교체가 환자에게 필요합니다.