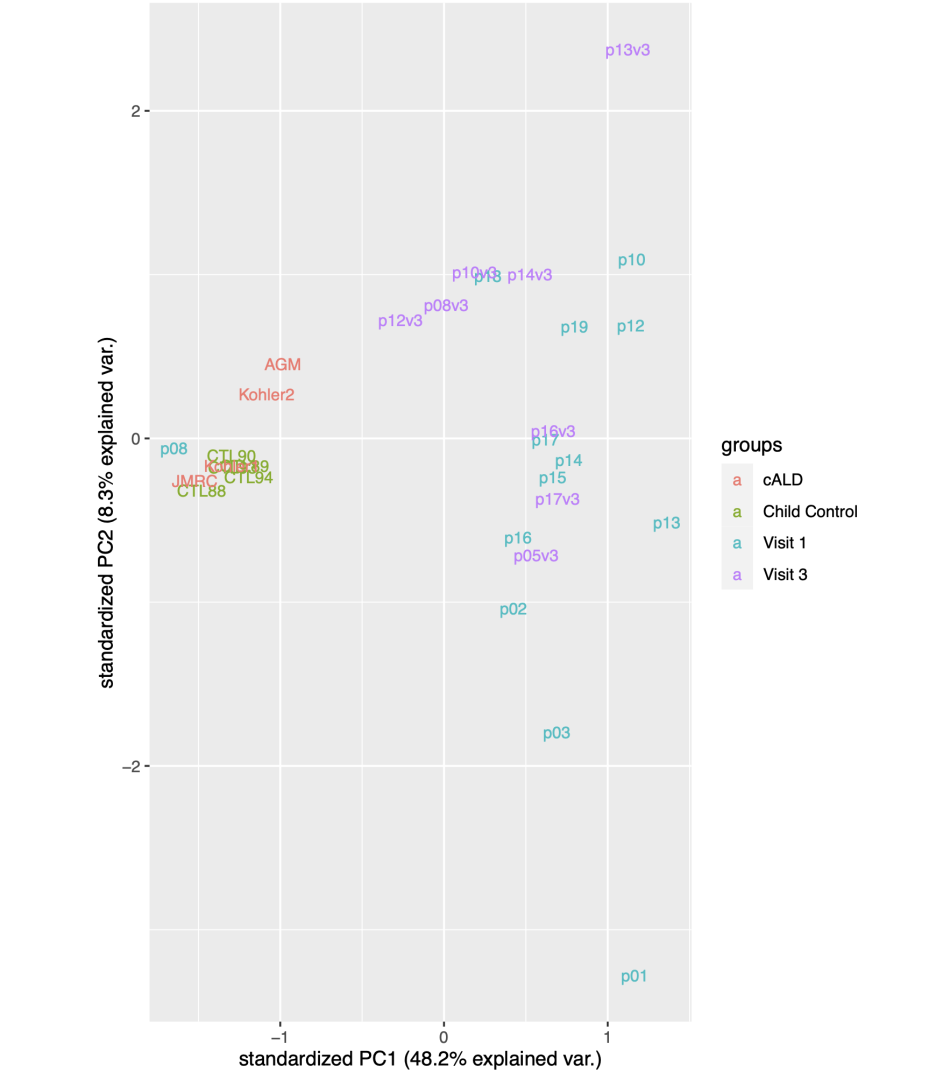
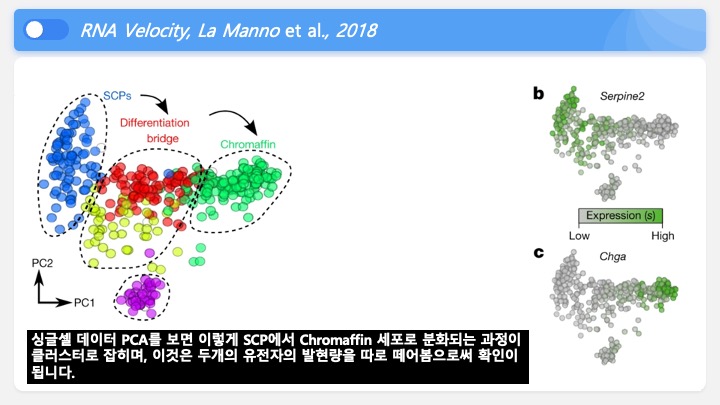
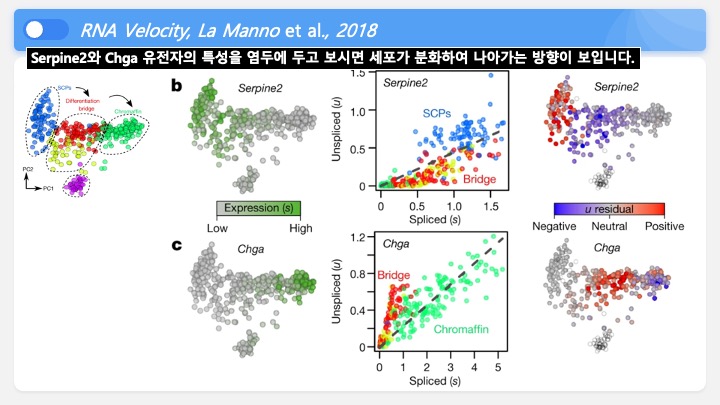
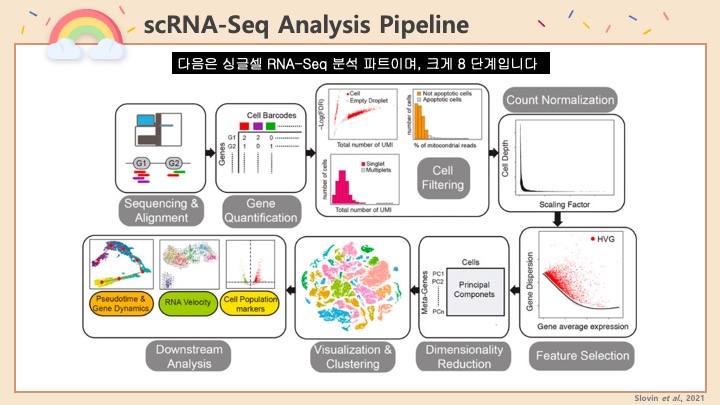
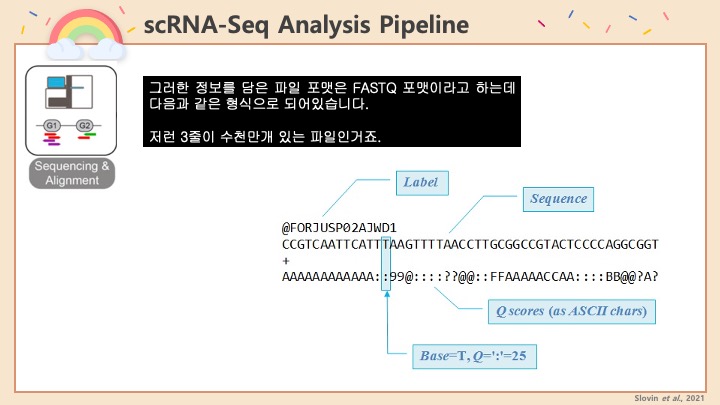
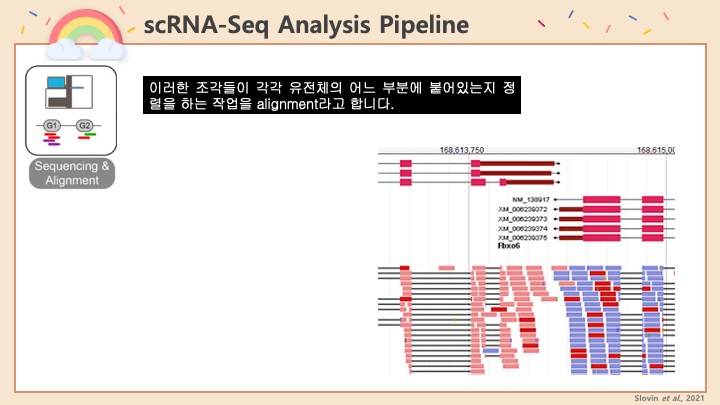
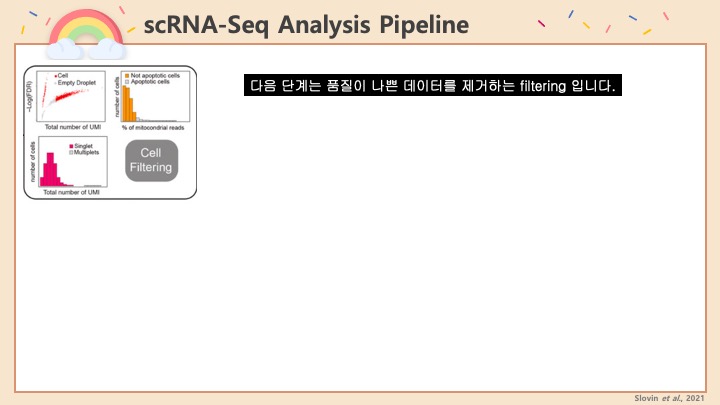
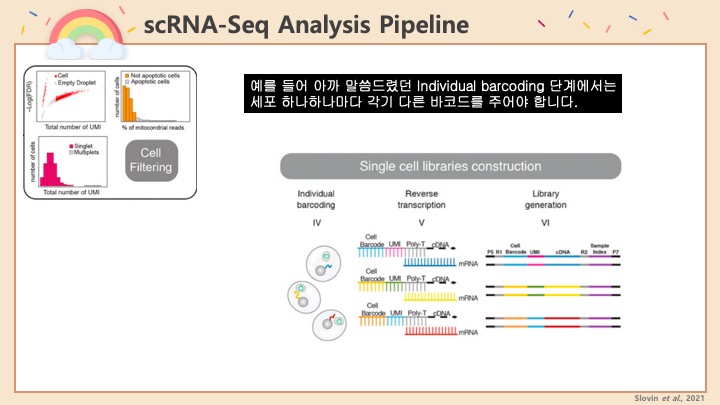
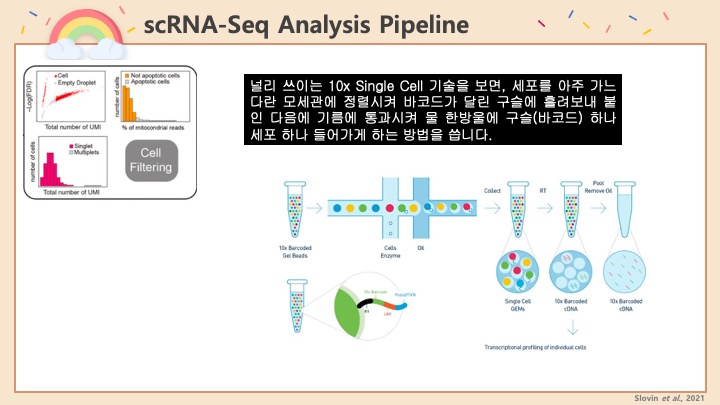
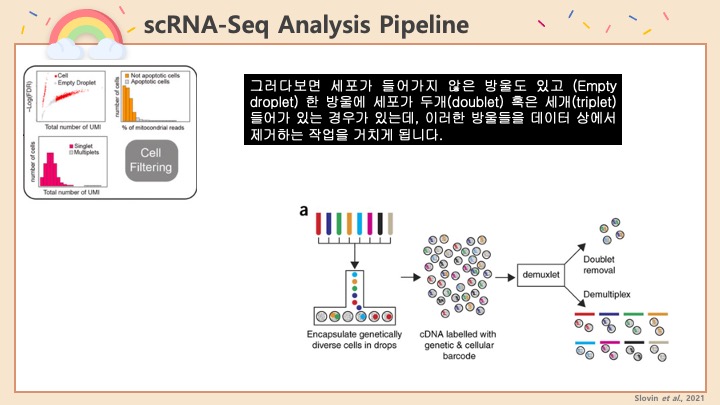
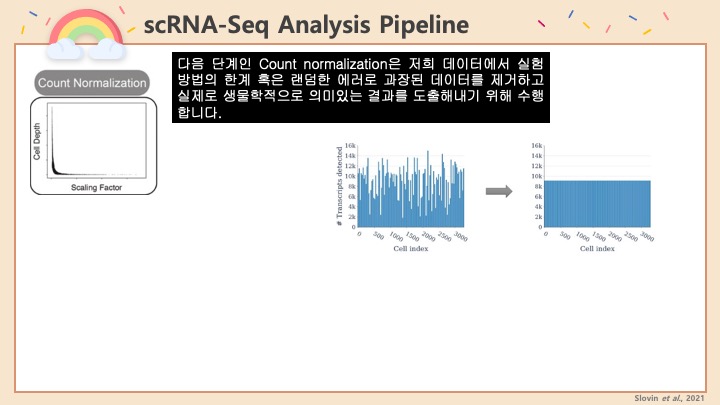
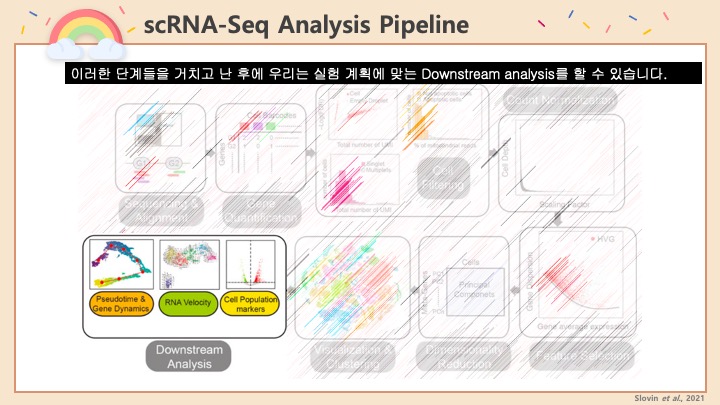
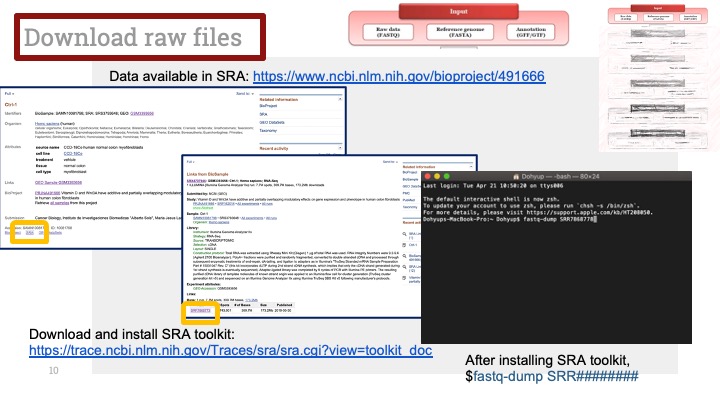
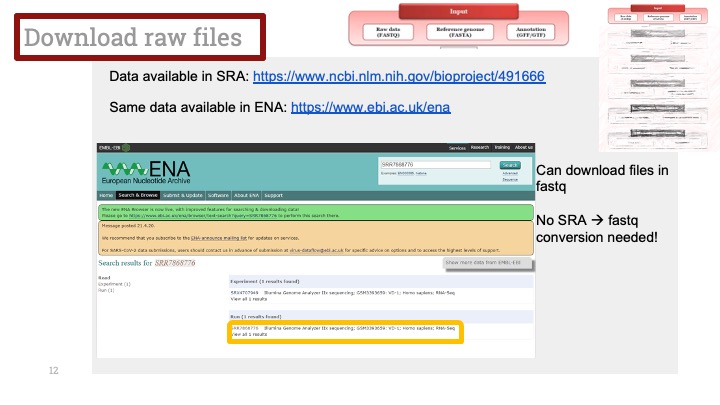
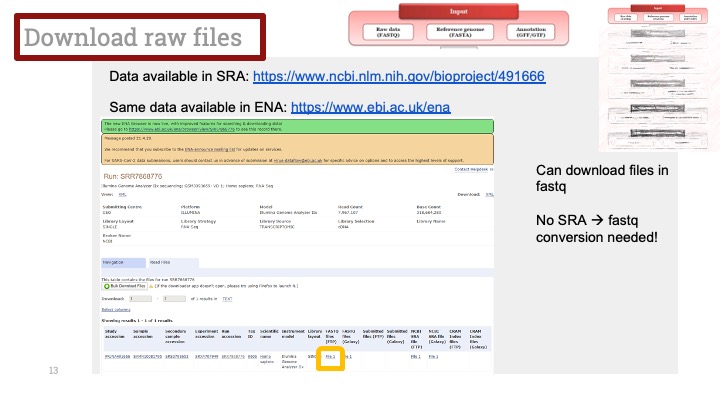
RNASeq, 특히 싱글셀 RNASeq 같은 고차원의 high-throughput 데이터들은 noise가 많이 들어올 수 있기 때문에, PCA로 한번에 딱! 보려고 하면 막 겹치고 예쁘게 나오지가 않아서 논문에 내려고 하면 음... condition 별로 구분이 가긴가지만 확연하게 딱! 구분이 가지가 않아요.
이럴 때 쓰라고 있는 건 아니지만, PCA와 비슷한 분석 방법으로 PLS-DA라는 것이 있습니다. Partial Least Squares-Discriminant Analysis의 줄임말으로써, PCA는 데이터의 variance를 최대한 유지하면서 dimension reduction을 하려고 하는 반면에, PLS-DA는 사전에 그룹에 관한 정보를 미리 줌으로써 covariance를 최대한 유지하면서 분석하는 방법입니다. 자세한 계산방법이나 차이점 및 장단점은 다음에 다뤄보기로 하고 여기서는 이 알고리즘을 이용하여 플랏을 만들어 보겠습니다.
아주 간단합니다. 저희가 PCA에서 만들었던 AverageExpression 값을 그대로 써서 Mixomics 패키지를 이용해서 그려보겠습니다.
일단 mixomics 패키지를 깔고 불러옵니다.
그런 다음 아까 우리가 janitor 패키지를 이용해 0값을 제거해서 저장한 mySCT2 변수를 그대로 이용해서 PLSDA를 계산하고 그려줍니다.
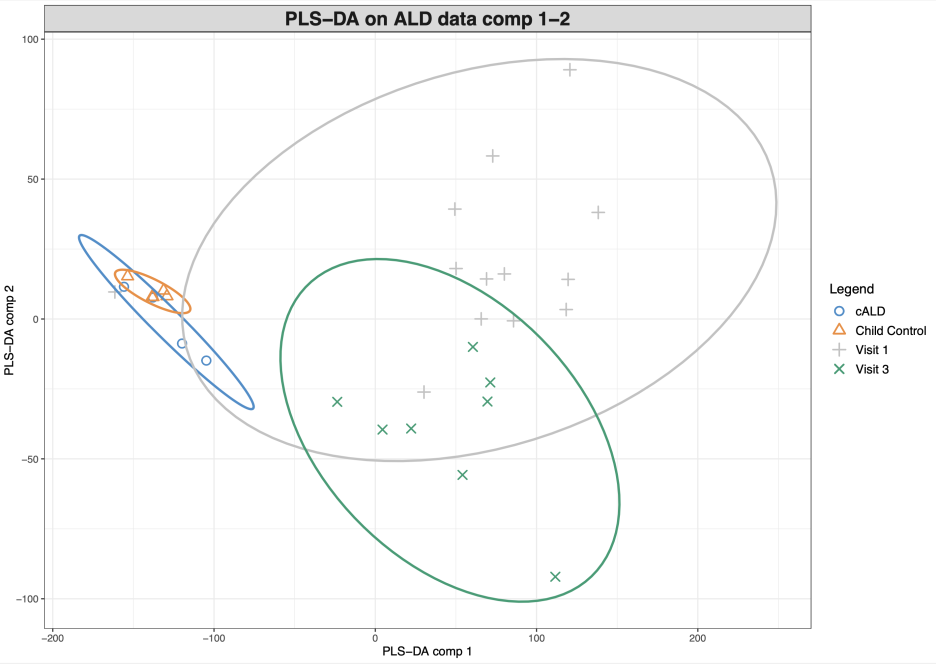
그러면 아래와 같은 플랏이 나와요.
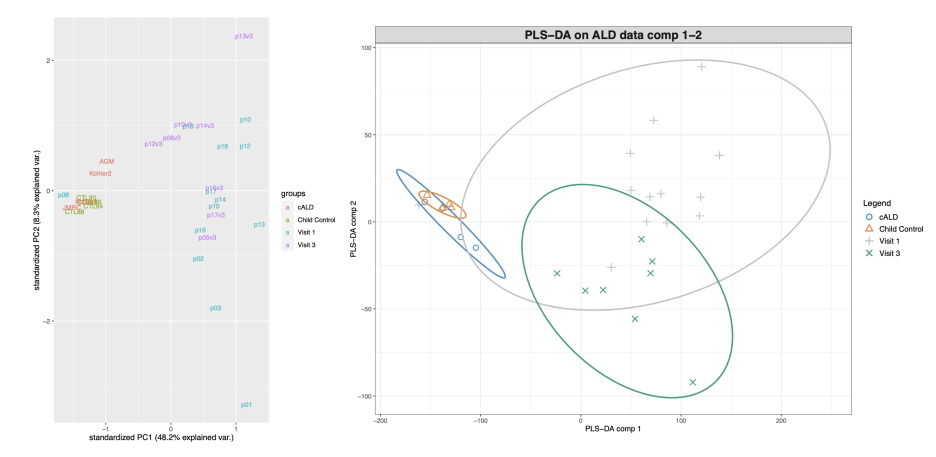
음... 아까 PCA랑 같이두고 비교해볼까요?
같은 데이터로 서로 다른 분석방법을 이용해 시각화를 했을 때에 왼쪽의 PCA랑 오른쪽의 PLS-DA랑 어떤게 condition간의 차이를 보기가 쉬울까요?
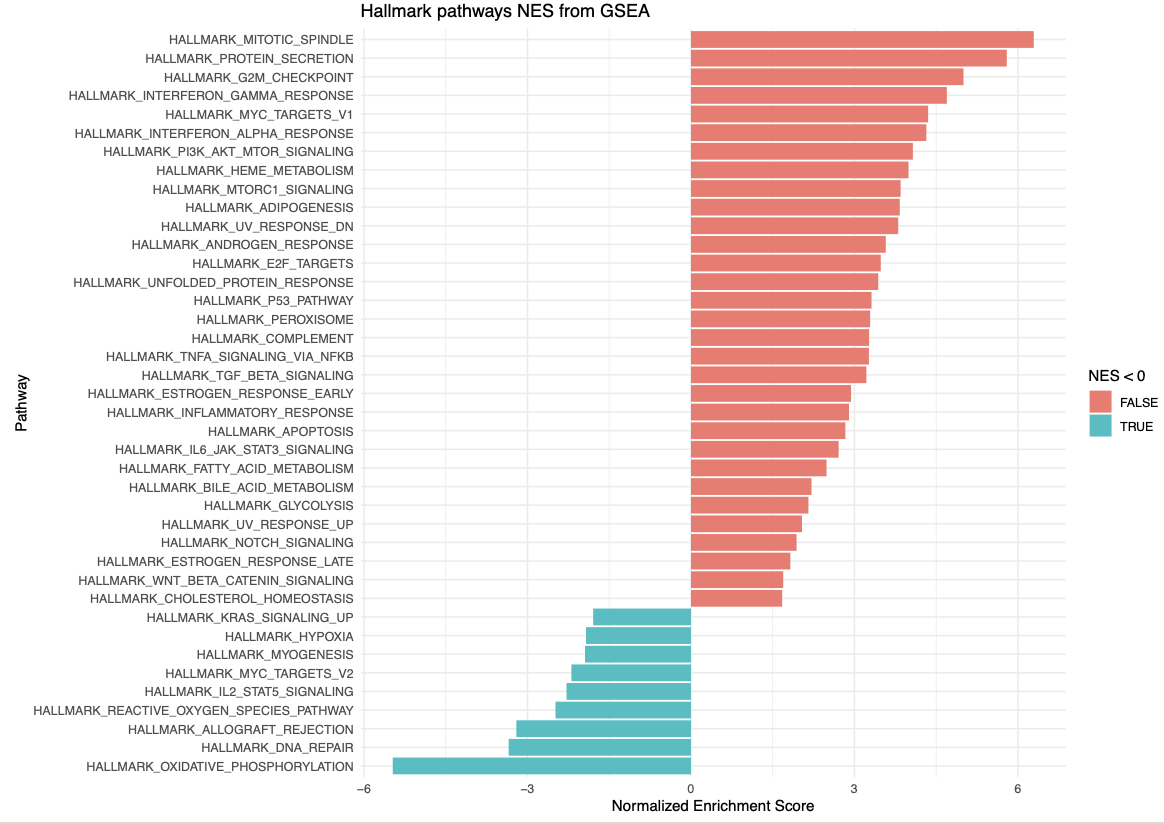
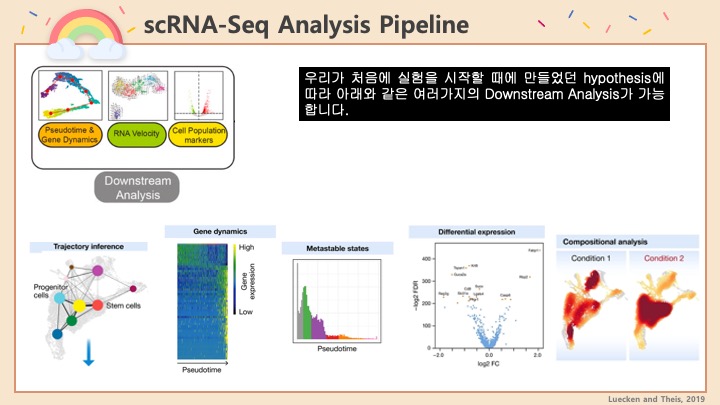
GSEA란 Gene Set Enrichment Analysis의 줄임말로 특정 유전자들의 집합이 있으면 이 유전자들이 어떠한 특성을 가지는 지 알아보는 분석 방법입니다. RNA 시퀀싱의 downstream analysis로 많이 쓰입니다. 간단하게 GSEA를 하는 방법은, 내가 원하는 Gene들을 p-value로든 Fold change로든 어떠한 점수를 기준으로 1위부터 꼴등까지 줄을 세웁니다. 그 후에 그 순위를 가지고 어떠어떠한 Gene Set에는 높은 순위의 유전자가 많이 들어있더라, 낮은 순위의 유전자가 많이 들어있더라 등을 확인합니다. 여기서 Gene set은 특정 pathway에 속하는 유전자들일 수도 있고, 공통된 기능을 가진 유전자 그룹일 수도 있고, 특정 질병에 노출되면 발현되는 유전자 리스트일 수도 있습니다. 사용자가 정하기 나름이죠. Broad Institute와 UC San Diego에서 이러한 유전자 세트, Gene set을 데이터베이스화 해서 정리해놓은 사이트가 있는데 그게 MsigDB입니다. 인간과 쥐의 유전자를 그룹별로 잘 정리해놓아서, GSEA에 유용하게 쓸 수 있습니다. https://www.gsea-msigdb.org/gsea/msigdb/
GSEA는 보통 그룹 간의 DEG (Differentially Expressed Gene), 즉 유의미하게 다르게 발현된 유전자들을 가지고 실행할 수가 있습니다. 이 때에 실행하는 GSEA는 내가 뽑은 DEG들이 어떠한 성격을 지니고 있는지 알아보는 것이겠죠. 예를 들어서, 두 환자 그룹에 한 그룹에는 비타민D를 1개월간 복용시키고 다른 그룹에는 플라시보를 복용시켜서 유전자 발현의 차이를 보는 경우에 두 그룹 간의 DEG를 구해 GSEA를 돌려 본다면 비타민D로 인하여 유의미하게 변한 유전자가 대체로 어떠한 그룹에 속하며 어떤 기능을 가지고 있는 지 알아볼 수 있습니다. 이 때, 이 DEG가 10개-20개 정도면 뭐 하나하나 살펴볼 수 있겠지만, 100개에서 500개 이렇게 나온다면 하나하나 보는 건 너무 많은 시간과 노력이 들겠죠. 그러면 GSEA가 이러한 시간을 아주 줄여줄 수 있습니다. 비슷하지만 다른 방법의 GSEA로는, DEG만 넣는 게 아니라 모든 유전자의 발현 정보를 넣어서 GSEA를 실행해볼 수도 있습니다. 이 때에는 입력 값이 아주 크겠지만, 위의 예제로 본다면 비타민D가 전반적으로 어떠한 영향을 미치고 어떤 pathway가 변화하는 지 종합적으로 알 수가 있습니다. 위의 두 방법 중에 어느 것이 옳고 어느 것이 틀리고 그러지는 않습니다. 다만, 내가 알아보려고 하는 질문에 따라서 전자 혹은 후자를 택하기도 합니다. 이 글에서는 Single cell RNA-Seq 분석을 할 때에 그 Downstream analysis로 Seurat 객체를 가지고 GSEA를 하는 방법을 간단히 기록하려고 합니다.
일단 먼저 Seurat을 불러오고 내가 이용하려는 데이터를 불러옵니다. 또한, 우리가 하려는 GSEA는 모든 유전자를 줄세워서 입력하는 위에 적은 후자의 방법을 쓰기 때문에, 유전자를 그룹 간에 비교한 값이 필요합니다. Seurat으로는 2만개 이상의 유전자를 모두 비교하는 데 엄청나게 오래 걸리기 때문에, presto 패키지에 있는 Wilcoxon rank sum test를 불러와서 사용하려고 합니다.
library(Seurat)
library(devtools)
install_github("immunogenomics/presto") #인스톨을 최초로 한 다음에는 이 줄은 생략합니다
library(presto)
#또 나중에 필요한 라이브러리 들을 여럿 불러와 보겠습니다.
library(msigdbr)
library(fgsea)
library(tidyr)
library(dplyr)
library(ggplot2)
library(tibble)
library(tidyverse)
library(data.table)
그런 다음, Wilcoxon rank sum test를 모든 유전자 대상으로 수행해줍니다. 제 Seurat 데이터는 myData3에 저장되어 있으며 저는 그 중 celltype.condition에서 특정 세포 (CD14+ Monocytes)의 질병 그룹(cALD)과 건강한 비교군 (Control) 간의 차이를 보고 싶습니다.
이제 앞서 말한 mSigDB에서 Gene Set들을 불러와야 합니다. 저는 가장 기본으로 유전자를 50개의 질병 및 생명 현상으로 나눈 Hallmark gene sets를 먼저 보려고 합니다. 그리고 이 불러온 Gene Set을 GSEA에 필요한 정보만 뽑아서 정리해 줍니다.
#msigdbr_show_species()
#위의 코드는 모든 가능한 생물 종을 표시해 줍니다.
#"H"로 Hallmark gene set을 불러 옵니다.
m_df<- msigdbr(species = "Homo sapiens", category = "H")
#불러온 Gene Set을 정리해 줍니다.
fgsea_sets<- m_df %>% split(x = .$gene_symbol, f = .$gs_name)
#불러온 Gene Set을 살펴봅니다.
head(m_df)
dplyr::count(myData.genes, group)
그런 다음에 제가 원하는 그룹을 뽑아서 logFC와 AUC score를 빼낸 뒤 정리해봅니다. 저는 CD14+ Mono_cALD 그룹에서 어떠한 pathway들이 더 혹은 덜 발현되는지 알아보고 싶습니다.
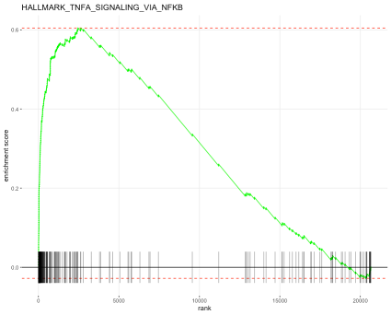
fgsea는 인풋으로 1) 미리 지정한 Gene Set 이 필요하구요 (m_df로 불러들여서 fgsea_sets로 포맷을 맞춤) 2) 유전자의 리스트와 각각 유전자의 순위 혹은 순위를 지정할 수 있는 점수가 필요합니다 (myData3에서 auc를 계산하여 myData.genes에 저장, 그런 다음 CD14Mono.genes에 순서대로 저장)