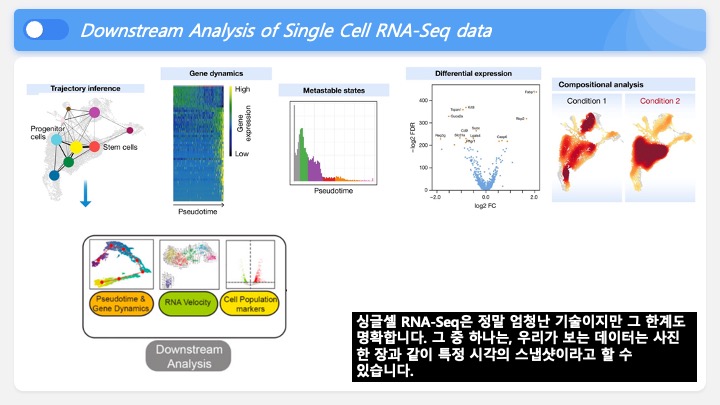
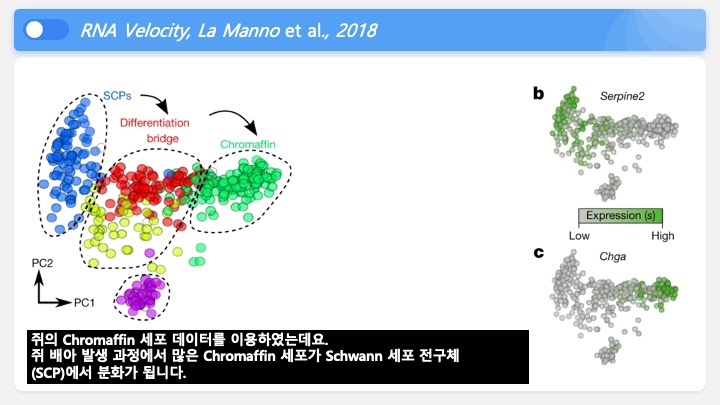
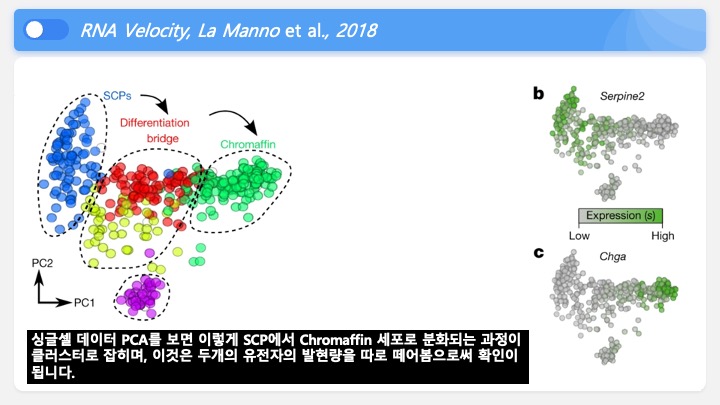
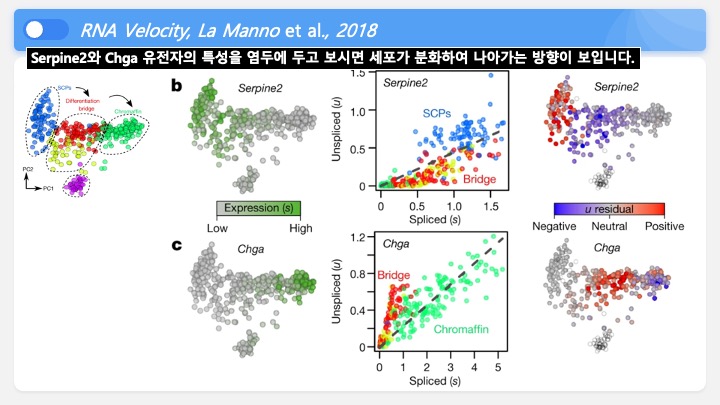
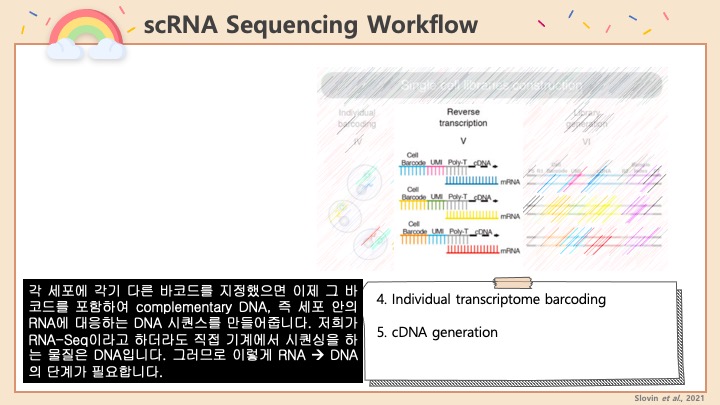
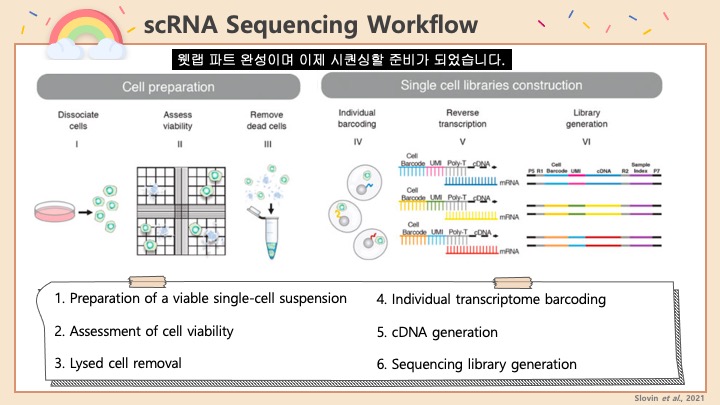
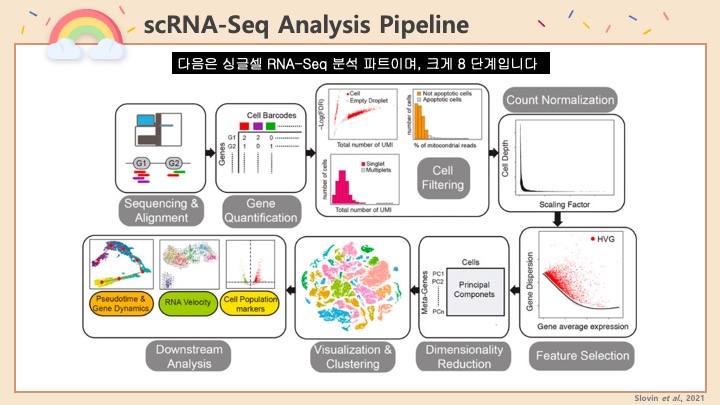
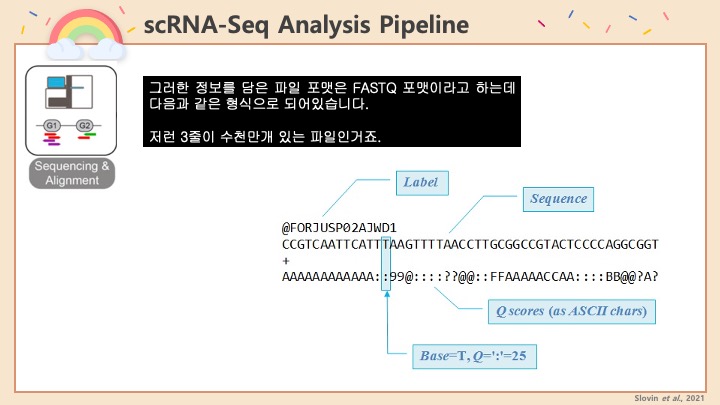
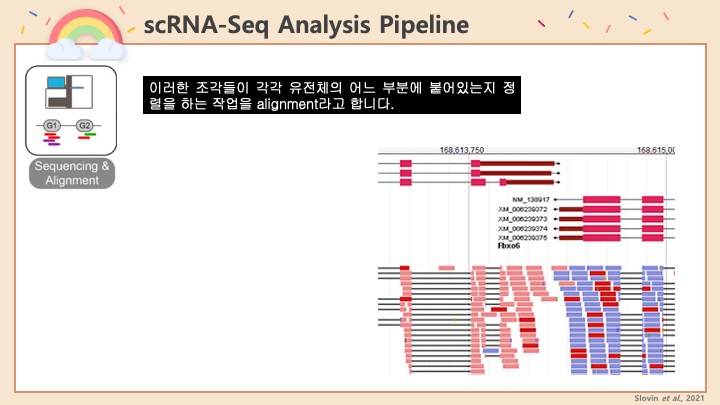
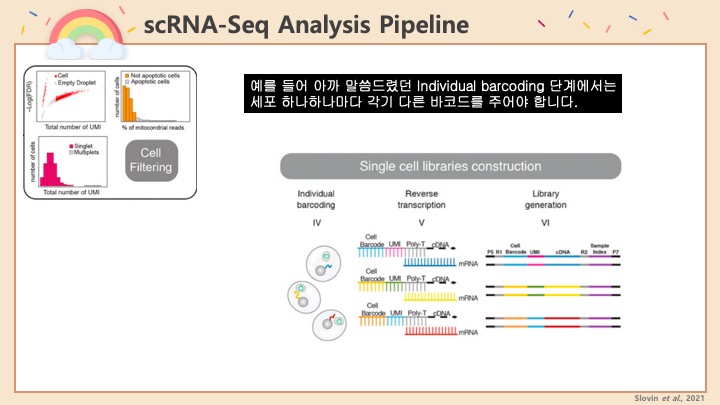
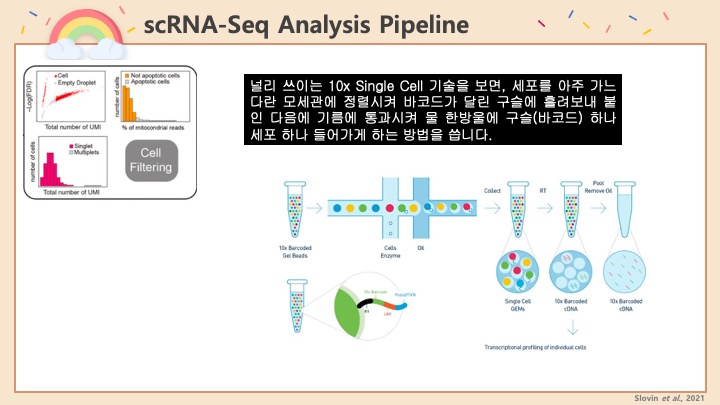
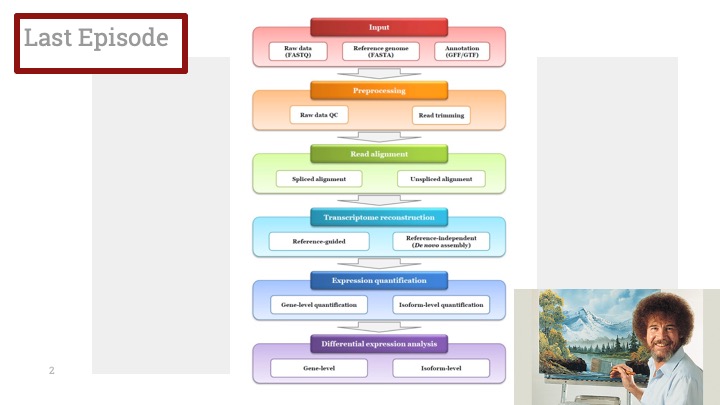
Single cell RNA-Seq 데이터를 전처리하는 과정에서 필수적인 단계 중 하나는 배치 효과 수정(batch effect correction)입니다. 하지만 이 단계에서 많이들 혼란스러워 하죠. 이 글에서는 싱글셀 알앤에이 시퀀싱의 배치 효과를 다룰 때에 가장 많이 올라오는 질문들에 관하여 다뤄보겠습니다.
What is Batch Effect in Single-cell RNA-Seq? 싱글셀 알앤에이 시퀀싱에서 배치 효과는 무엇인가요?
Batch effect는 샘플 그룹에서의 차이가 생물학적인 요인이 아니라 기술적인 방법에서 와서, 잘못된 결론을 도출되도록 할 때에 일어납니다. 이 시점에서, batch effect를 수정해야할 필요성이 생기는 거죠.Batch effect correction은 서로 다른 연구나 실험, 혹은 실험 과정에서 온 세포들 혹은 샘플들을 합칠 때에 생기는 기술적인 차이를 제거하는 것입니다.
그거 정규화(normalization)이랑 엄청 비슷하게 들리는데요?
기억해 둘 점: scRNA-Seq normalization도 기술적인 노이즈나 치우침을 제거하는 것을 목표로 데이터에서 나온 유전자 표현의 차이가 정말로 생물학적인 차이에서 오도록 하는 것입니다.
Normalization과 Batch effect correction이 바로잡는 기술적인 노이즈에는 다른 점이 있습니다.
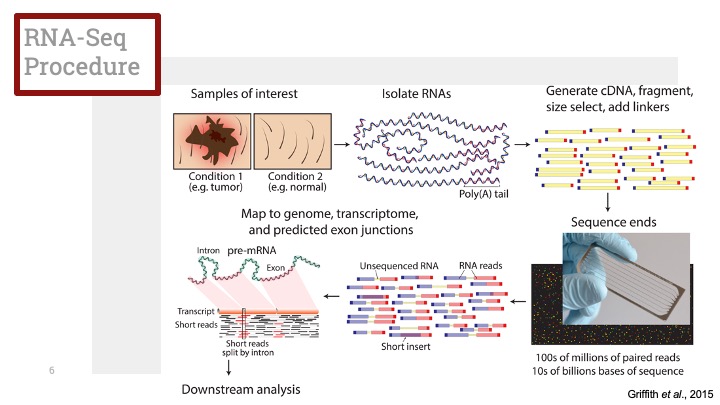
- Normalization은 라이브러리 제작, 대규모의 dropout, 유전자 길이 및 GC 비율 등에 따른 증폭 치우침 (amplification bias) 등이 타겟입니다. (Jiaet al., 2017)
- Batch effect correction은 실험 설계 및 실제 실험 수행 (시퀀싱 기계, 실험 시각, 시약, 실험실 등)에서 오는 변이를 통제하는 것이 타겟입니다 (Haghverdiet al., 2018)
또 하나의 다른 점은 입력하는 데이터입니다. 특히, normalization은 raw count matrix (예를 들어 세포 x 유전자)를 입력값으로 받지만, batch effect를 제거하는 대부분의 방식은 차원이 축소된 데이터 (dimensionality-reduced data; PCA에서 첫 50개의 PC)를 이용하여 계산 시간을 줄입니다. 이것은batch effect가 제거된 결과가 시각화나 그래프 기반의 클러스터링에는 유용하지만, DEG를 찾는 등의 그 후 다른 분석에서는 축소 전의 원래 데이터가 요구됨을 의미합니다. 이러한 방식의 예외도 있는데 raw count table을 사용하는 Mutual Nearest Neighor (MNN) 및 scGen 등의 메소드는 normalization이 된 gene expression matrix를 결과값으로 배출합니다 (Tranet al., 2020).
How to detect batch effect in single-cell RNA-Seq? 싱글셀 RNASeq에서 배치 효과는 어떻게 알아내나요?
Batch effect를 식별하는 전용 툴들이 있기는 하지만, 다음의 간단한 테스트로도 충분합니다.
1. Dissect Principal Components 주성분분석 뜯어보기
주성분분석(PCA; Principal component analysis)는 어떠한 인자가 데이터에서 가장 변이가 큰 지 밝혀줘서, batch effect의 좋은 지표가 됩니다.
Raw data에 주성분분석을 실행한 다음, 상위 주성분(PCs)들을 살펴봅니다. 주요한 변이가 생물학적인 차이보다 실험 batch에 따라간다면, batch effect가 존재한다는 것을 알 수 있습니다.
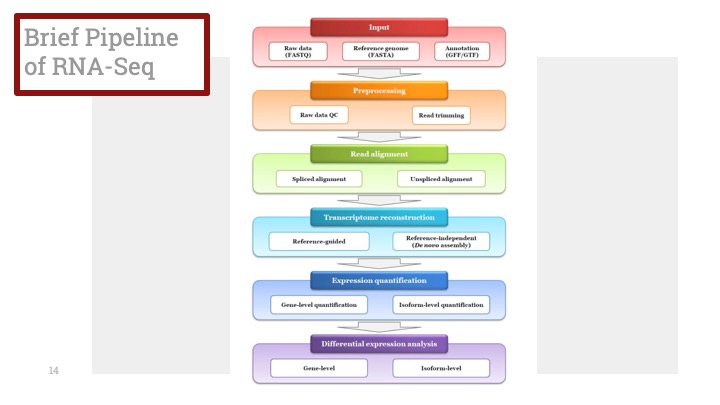
2. Examine Clusters 클러스터를 살펴보기
PCA와 비슷하지만, 시작적으로 더 이해하기 쉬운 클러스터링 분석도 또한 batch effect를 발견하는 데에 도움이 됩니다. 간단히 클러스터링 분석을 실행한 후에 t-SNE 혹은 UMAP 그래프를 batch correction 전/후로 비교해 봅니다. 그 원리는, 만약 batch effect가 존재 하고 수정되지 않은 채로 있으면, 생물학적인 변이 때문이 아니라 서로 다른 batch들 간의 세포들이 클러스터 하는 것 보입니다. batch correction을 한 다음에는, 그러한 것들은 클러스터링에서 사라집니다.
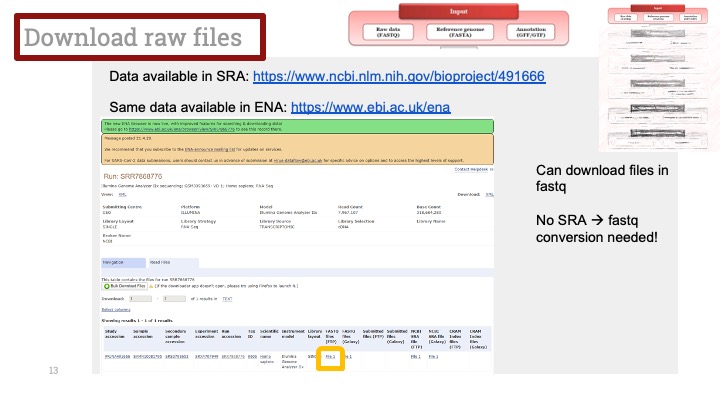
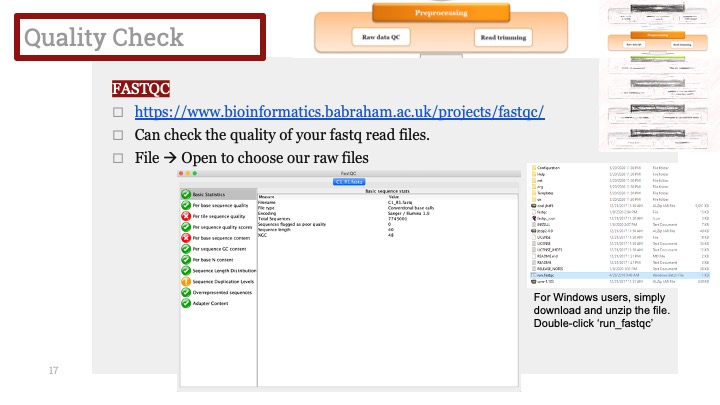
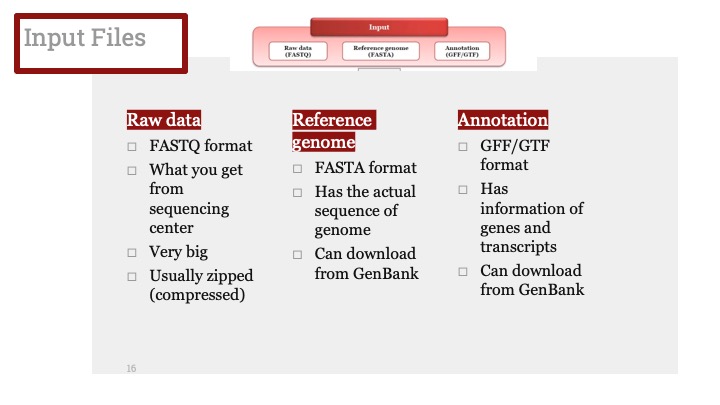
아래는 말초혈단핵세포(PBMC; Pheripheral blood mononuclear cells) 샘플에서 나온 Kang et al. (2018)의 데이터입니다. 이 데이터는 두개의 batch가 있습니다. 하나는 전신성홍반루푸스(SLE; systemic lupus erythematosus) 환자 샘플이고 (batch 1), 다른 하나는 컨트롤입니다 (batch 2). Batch correction 없이는 클러스터들이 완전히 batch에 따라 분리가 됩니다 (Figure 1). 하지만 다행히도 MNN(Mutual nearest neighbor) 알고리즘을 사용하여 batch correction을 하고 난 뒤에는 세포 클러스터링에서 batch effect를 찾아볼 수가 없습니다 (Figure 2).
Figure 1. t-SNE plot for PMBCs (Kang et al., 2018 ) without batch effect correction. The clusters are color coded by batches: blue = batch 1 (patients), red = batch 2 (controls). Data processed and visualized by BBrowse r
Figure 2. t-SNE plot for PMBCs (Kang et al., 2018) , batch effect corrected by MNN. The clusters are color coded by batches: blue = batch 1 (patients), red = batch 2 (controls). Data processed and visualized by BBrowse r
How do you deal with batch effect in single-cell RNA-Seq? 배치 효과는 어떻게 예방하나요?
1. Prevent potential batch effect 잠재적인 배치 효과의 가능성을 제거하세요
Batch effect가 실험 디자인과 실행 단계에서 생성되기 때문에, 가장 쉬운 해결방법은 탄탄한 실험 계획을 만드는 겁니다! 그러나 가장 이상적인 실험 세팅도 batch effect를 없애기 보단 줄이기만할 수 있으니까, 기술적인 추가 단계가 개발되고 있습니다.
그 예 중 하나는 Cell Hashing입니다. Cell hashing은 많이 번역되는 세포 표면 단백질에 붙는 항체에 짧은 DNA 시퀀스를 붙여 각각의 세포에 바코딩을하는 방법입니다. 다른 예시 중 하나는 Spike-in 인데요. 이것은 유전자 발현량을 정규화하는데에 도움을 줍니다. Spike-in은 시퀀싱 전에 샘플에 첨가하는 인공적으로 합성된 RNA 서열이며, 이 후 분석에서 쉽게 구분이 가능합니다. 이러한 spike-in의 양을 측정하면 샘플 간의 변이를 효율적으로 통제하고 줄일 수 있습니다.
2. Choose a batch effect correction algorighm 배치 효과를 수정하는 알고리즘을 선택하세요
이제까지 여러가지의 batch effect 제거 알고리즘이 개발되어 왔습니다. 그리고 거기에 관한 리뷰도 여러가지 있습니다. 하나 추천하는 리뷰 논문은 Chen et al. (2021) 입니다.
각각의 알고리즘은 그 목표에 어떻게 도달하는 지가 다른데, 여기서는 대표적인 세가지의 알고리즘을 간단히 살펴보겠습니다. 이 세가지 알고리즘은 싱글셀 시퀀싱 데이터의 특징인 많은 수의 세포와 높은 drop-out을 염두에 두고 만들어졌습니다. 이것은 bulk RNA-Seq 및 microarray 데이터를 위해 개발된 기존의 알고리즘보다 더 성능이 뛰어나게 만들어줍니다.
Mutual Nearest Neighbors (MNN) 알고리즘. (Haghberdi et al., 2018) 이름에서 볼 수 있듯이, 이 알고리즘은 batch들 사이에서 가장 비슷한 세포들을 찾아내는 것이 목표입니다. 이러한 세포들은 mutual neighbor로 간주되며, 알고리즘은 그것들이 같은 세포 유형이라고 추정합니다. 그러므로 그것들 사이에서의 차이점은 batch effect 때문에 생긴 것이겠죠. 이러한 차이점의 정도는 batch effect가 얼마나 강한지를 보여줍니다. 이 차이점을 수식화하여 그 정도에 따라 batch들을 합칠 때에 이용합니다.
Seurat Canonical Correlation Analysis (CCA) (Butler et al., 2018) 이 알고리즘은 공통적으로 연결된 구조 (혹은 canonical correlation vectors)를 찾습니다. 이러한 벡터들이 batch를 서로 합칠 때에 세포들을 정렬하도록 도와줍니다.
Harmony (Korsunsky et al., 2019) 이 알고리즘은 먼저 주성분분석을 하여 세포를 저 낮은 차원 공간에 깔아줍니다. 그런 다음 각각의 클러스터의 고유한 수정 요소(correction factor)에 기반하여 그 클러스터의 중심을 찾아냅니다. 그리고 세포들은 그 수정값에 의해 재배열됩니다. 이 과정들이 batch effect가 제거되고 클러스터들이 완벽하게 겹쳐질 때까지 반복됩니다.
위의 세가지 알고리즘을 포함한 11가지의 batch effect correction 메소드들은 Tran et al. (2020) 에 잘 리뷰되어 있습니다. 이 리뷰 논문에서는 같은 세포 유형이지만 다른 기술이 쓰인 경우, 서로 다른 유형의 세포인 경우, batch가 여러번인 경우, 아주 큰 데이터인 경우, 시뮬레이션으로 생성된 데이터인 경우 등의 다섯가지 시나리오에서 분석되어 있습니다. 각각의 경우에서 제일 효과적인 알고리즘은 다르지만, 전반적으로 저자는 Harmony와 Seurat CCA를 추천했으며, Harmony의 수행 시간이 더 빨랐기 때문에 그걸 더 추천했습니다.
3. Check for Overcorrection 과도한 수정인지 확인하기
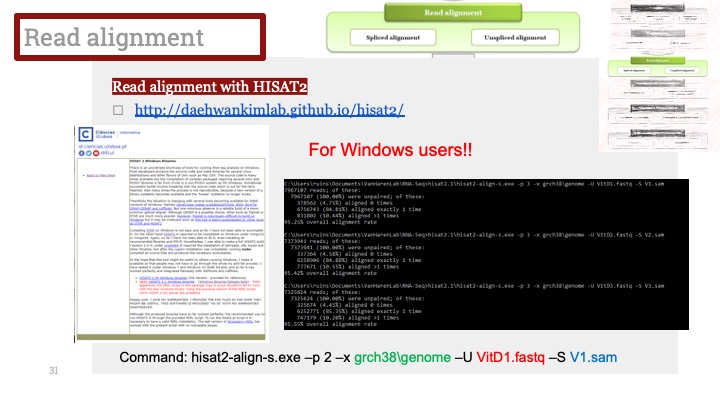
다른 전처리 단계와 마찬가지로, batch effect correction은 과도하게 수행될 수도 있습니다. 이러한 일은 알고리즘이 생물학적인 차이를 batch effect로 잘못 인식하고 그걸 제거해버릴 때에 생겨납니다.
내 데이터에서 과도한 수정(overcorrection)인지 어떻게 알 수 있을까요? 가장 명백한 사인은 세포들이 아주 많이 겹치는 것입니다. Figures 3 and 4를 보면 과도하게 수정된 데이터셋을 관찰할 수 있습니다. Neuroal ceroid lipofuscinoses (NCL) 단백질, Chromogranin A, 및 parathyroid secretory 단백질을 각각 따로 혹은 동시에 발현하는 세포에는 3가지 유형이 있습니다. Batch effect correction을 적용하고 나니 batch들이 완전히 겹쳐서 세포 유형을 구분할 수 없게 되었습니다 (Figure 3).
Figure 3. Batch effect overcorrection으로 인해 세포 유형이 모두 겹쳐버렸습니다.Figure 4. batch effect correcion을 없앴더니 세포 유형별로 클러스터가 나눠졌습니다.
이러한 경우, 데이터에 더 잘 맞거나 덜 강력한 수정 알고리즘을 적용해봅니다. 또한, batch correction이 과연 필요한가도 고려해볼 수 있습니다. 만약 PCA 상으로 batch effect가 거의 보이지 않는 경우에는 특히 더 말이죠.
또한, batch effect의 유무와 수정한 결과를 평가하는 툴들도 개발되어 있습니다. 분석 파이프라인이 복잡해지긴 하지만, 이러한 툴들은 batch effect 제거가 필요한 경우에는 돌려보는 것이 좋죠. 이러한 툴에는, Buttner et al. (2019)의 kBET (Figure 5) 혹은 Korsunsky et al. (2019)의 LISI(local inverse Simpson's index) 등이 있습니다. 각각의 툴에는 장단점이 있습니다. 예를 들어 만약 자신의 데이터가 아주 다른 여러 세포 유형을 지니고 있다면, kBET보다는 LISI를 추천합니다. 반면에 LISI는 batch 들이 서로 다른 크기일 때는 결과가 좋지 않습니다.
Closing 마치며
결과적으로, 어떤 것이 제거되어야 할 노이즈이고, 어떤 것이 생물학적으로 흥미로운 결과인지는 연구자들이 살펴보려는 질문에 달려 있습니다. 예를 들어서, 같은 질병을 가진 남성과 여성으로 이루어진 연구는 종종 성별에 따른 클러스터링이 나오게 됩니다. 이러한 경우, 연구자들은 성별을 batch effect로 간주하고 성별을 제외한 시그널을 관찰해 볼 수 있습니다. 하지만 역시 성별과 병의 유무를 함께 보는 것도 도움이 되겠죠. 이러한 것들을 잘 파악하여 올바른 결과를 도출하기 바랍니다.