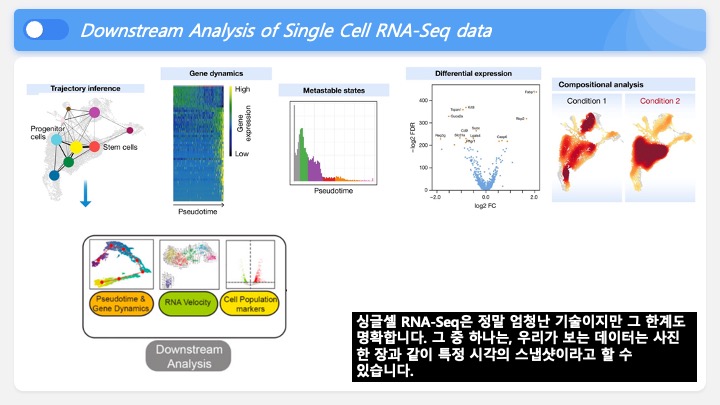
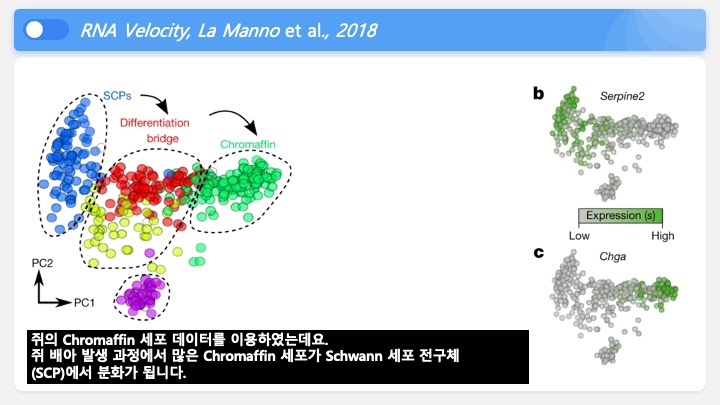
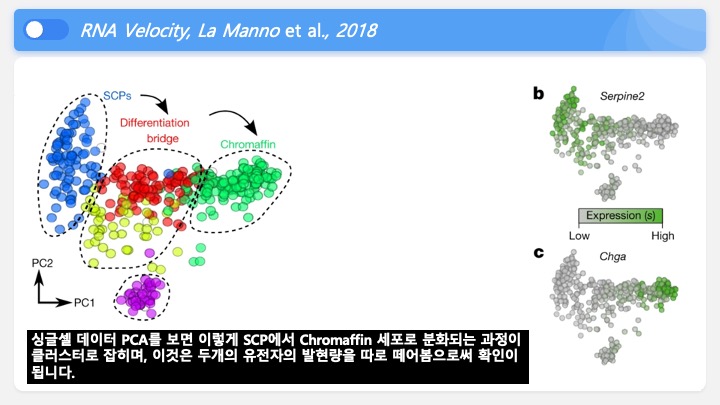
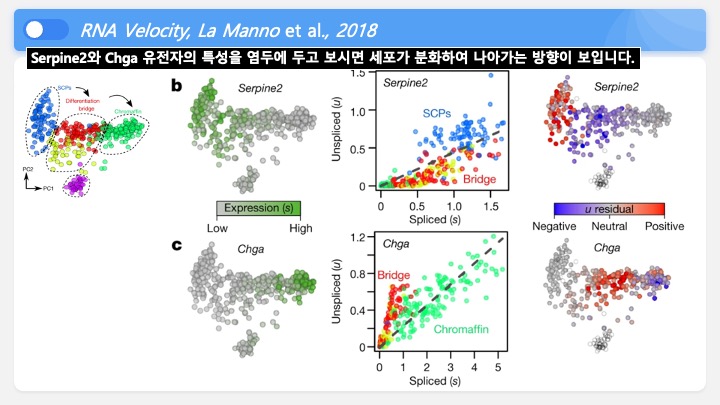
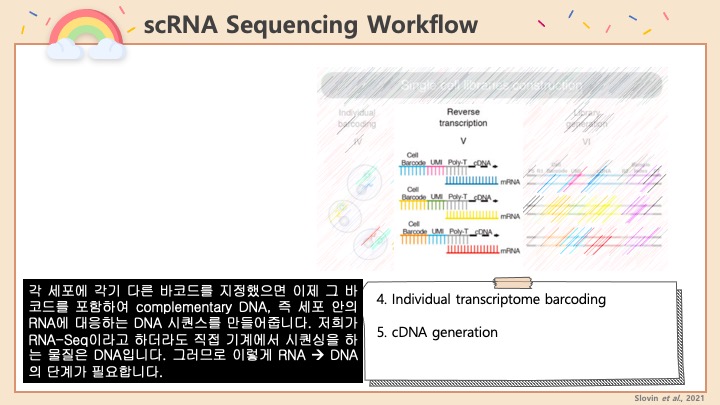
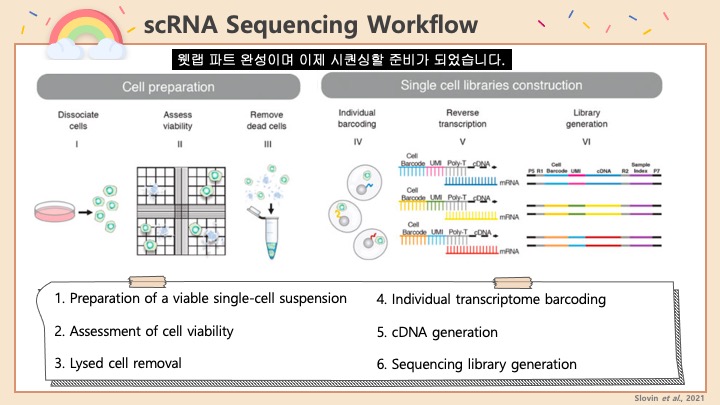
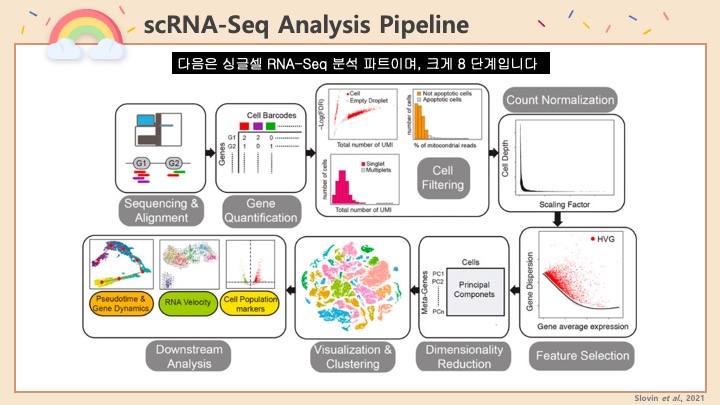
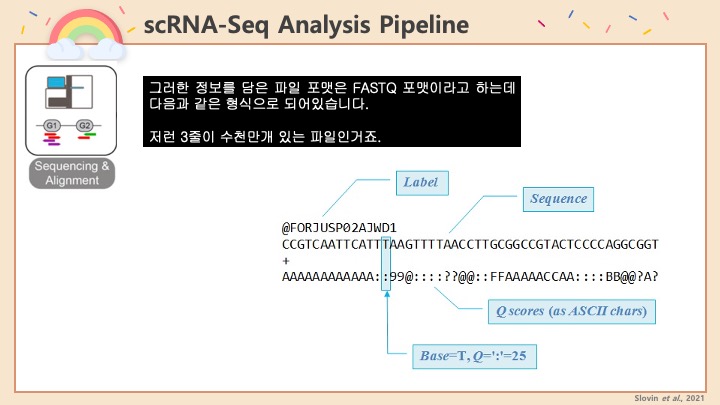
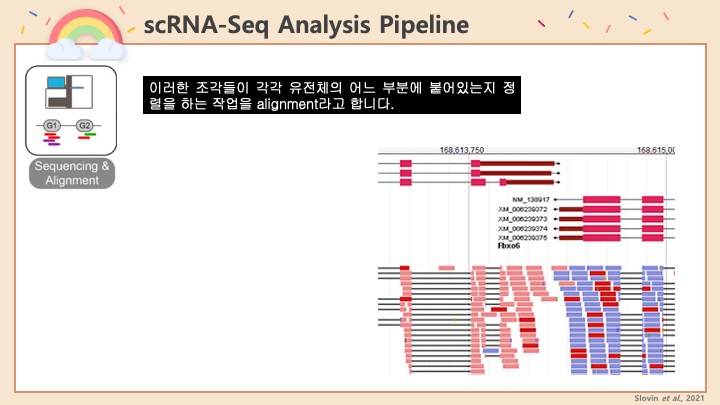
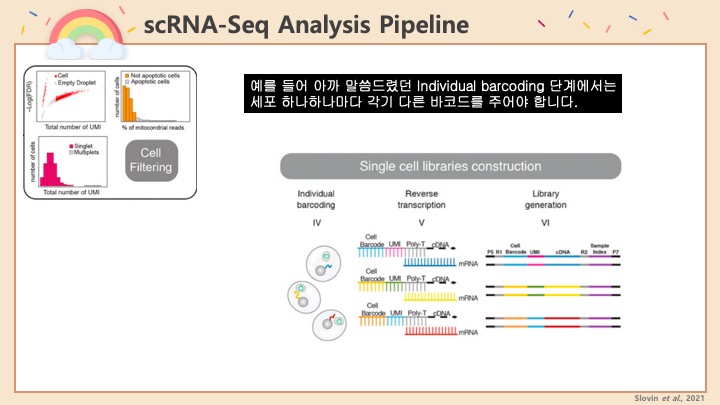
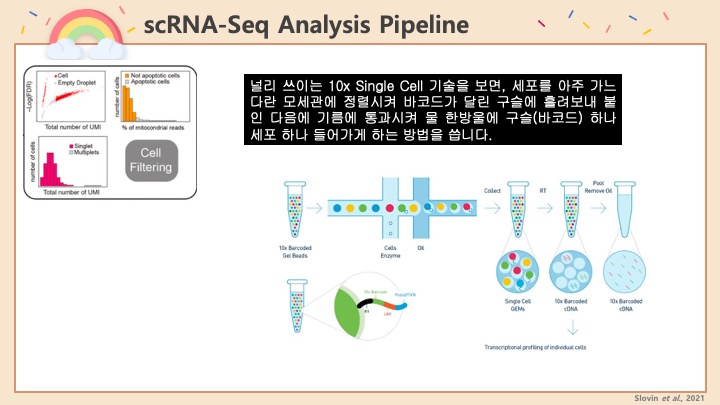
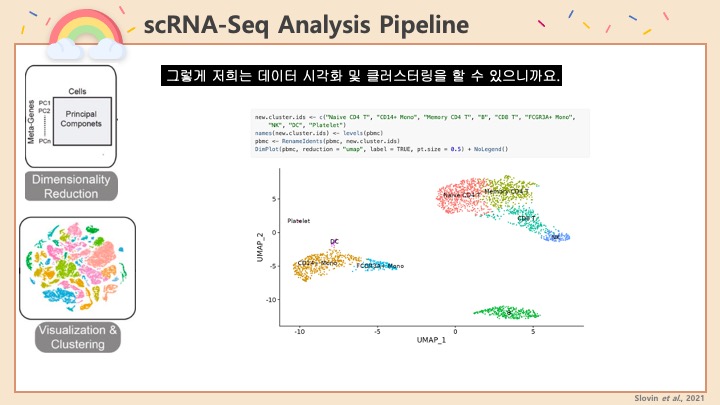
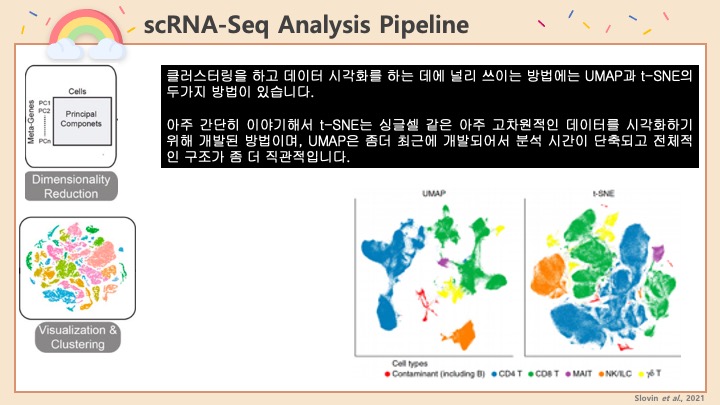
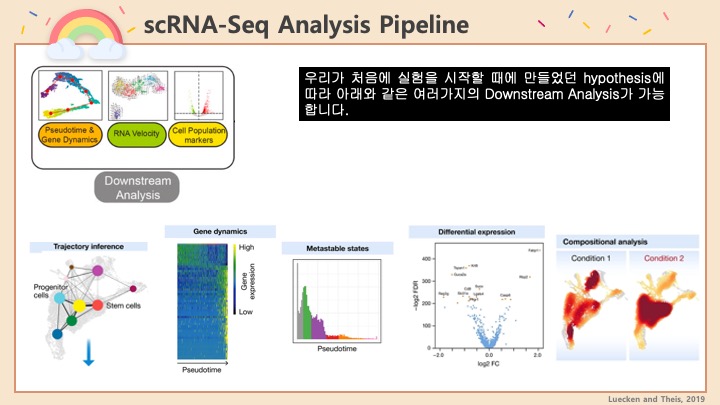
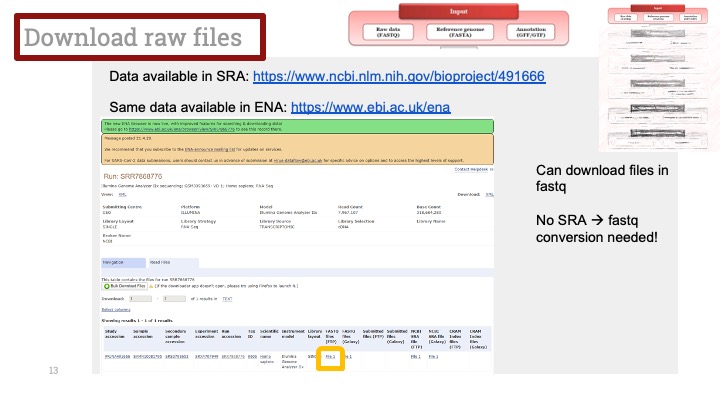
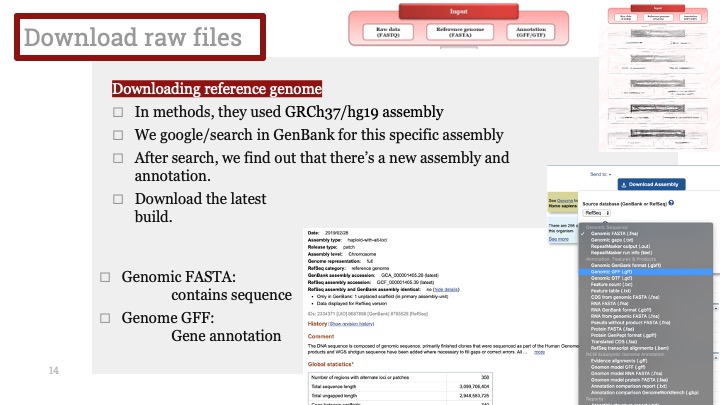
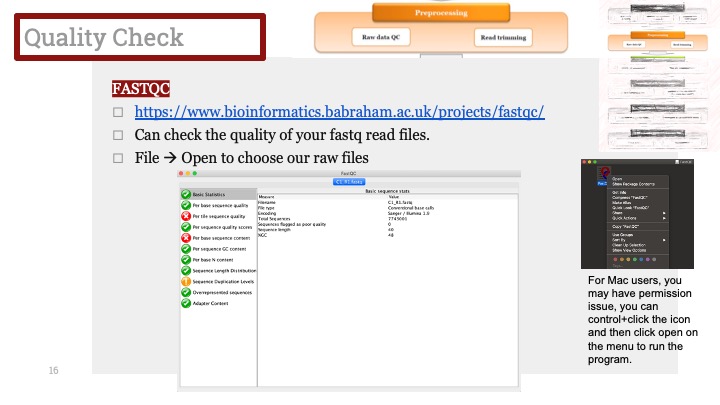
싱글셀 시퀀싱, scRNA-Seq 분석을 하다보면 가장 험난하고 어려운 단계가 각각 클러스터의 세포 종류를 지정하는 cell type annotation 인 것 같습니다. 저도 분석을 하면서 여러가지 방법을 시도해 봤는데, 그 기록을 남깁니다.
1. Seurat FindMarkers
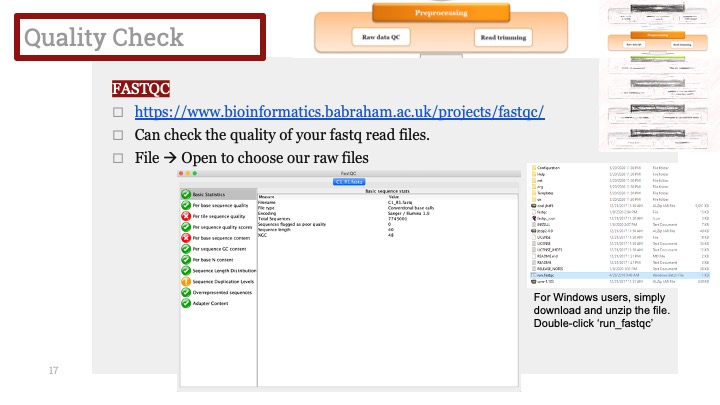
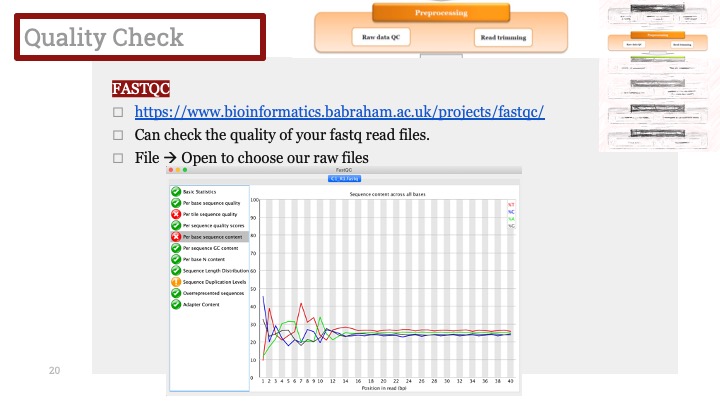
Seurat에서 제공하는 가장 기본적인 메소드인 FindAllMarkers를 이용하면 각각 클러스터에서 어떠한 유전자가 많이 발현되는 지 알 수 있습니다. 그것을 이용하여 클러스터에서 마커들을 찾고 데이터베이스 혹은 논문에 비교해봅니다.
장점: 간단하다.
단점: 수동 비교 및 결정이라 시간이 오래 걸리고 헷갈릴 때가 많다

Seurat tutorial에서 제공하는 코드입니다. 이 코드를 실행시키면, 각각의 클러스터에서 다른 세포들보다 과발현된 유전자들을 정리해줍니다. 저는 주로 이 결과를 csv로 정리해서 엑셀로 열어본 다음 여러가지 데이터베이스에 넣어보기도 하고, 논문을 찾아보기도 합니다.
주로 찾아보는 데이터베이스:
a. PanglaoDB: 싱글셀 데이터가 방대하게 모아져있으며 유전자를 넣으면 그 유전자가 어느 세포에서 얼마나 발현되는지 알려줍니다.
https://panglaodb.se/index.html
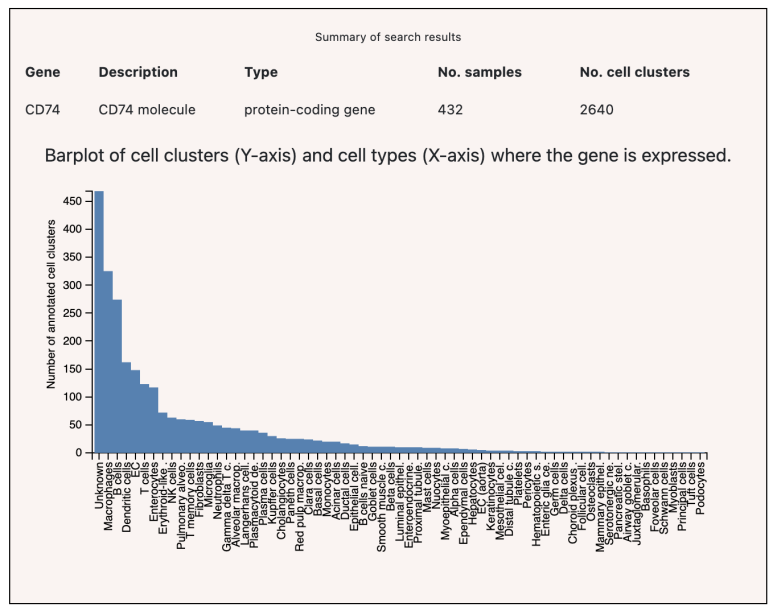
그리고 각 세포 종류마다 마커들을 모아놓은 페이지도 있습니다. 한가지 단점은 유전자를 하나하나 찾아보고 정리해야 한다는게 번거로울 때가 있습니다.
b. Single Cell Portal: 싱글셀 데이터를 모아 놓은 Broad Institute의 데이터베이스입니다.
이 데이터베이스의 장점은 여러가지 마커들이 앞의 FindAllMarkers로 나올 때에 한번에 입력하여 DotPlot으로 보여준다는 것입니다. 아주 편리하고 강력합니다.
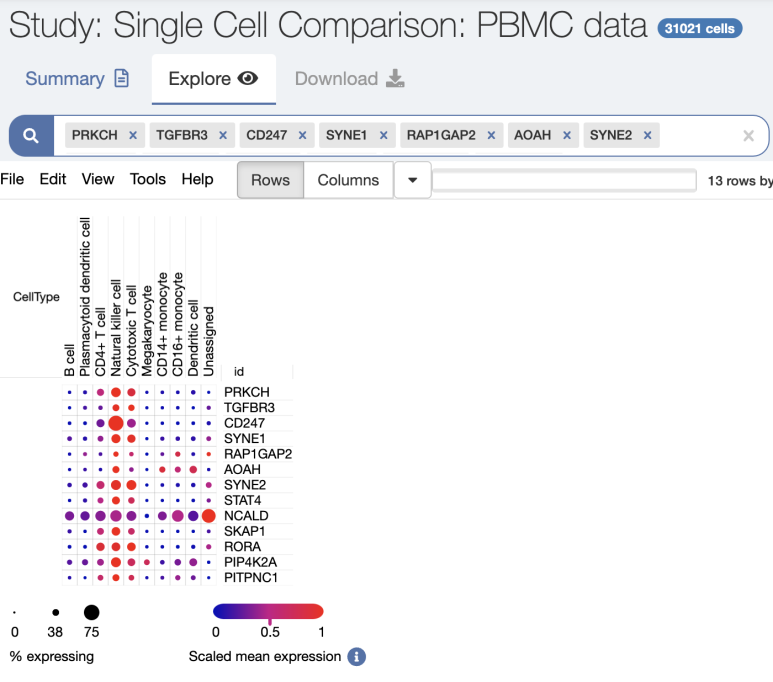
하지만 때때로 세포 종류의 resolution이 그리 높지 않습니다. 예를 들어서 B cell 중에서도 Memory B cell인지, Naive B cell인지 구분하고 싶을 때가 있는데 그렇게까지 구분은 해주질 않습니다.
c. 그외 논문이나 사이트: 특정한 세포의 subtype들을 구분하고 싶을 때에는 논문을 서치하는 게 빠를 수도 있습니다. 그게 아니라면 데이터베이스는 아니지만 저에게 유용한 사이트가 아래에 있습니다.
https://www.biocompare.com/Editorial-Articles/576304-A-Guide-to-NK-Cell-Markers/
Biocompare라는 사이트인데 거기서 올린 글에 유용한 정보가 많이 있을 때가 있습니다. 위의 링크는 NK 세포의 subtype에 관한 글이고 그 외에 T cells, B cells 에 관련한 글도 이 사이트에 있어서 도움이 많이 되었습니다.
2. SingleR
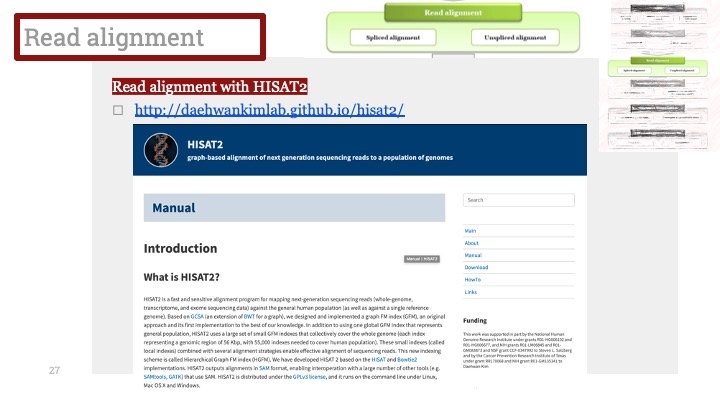
싱글알은 많은 연구자들이 쓰시는 Annotation 도구입니다. Bioconductor로 설치 가능하며, Seurat 오브젝트와 직접적인 호환은 되지 않지만, SingleCellExperiment로 변환하여서 간편히 실행할 수 있습니다.
https://bioconductor.org/packages/release/bioc/html/SingleR.html
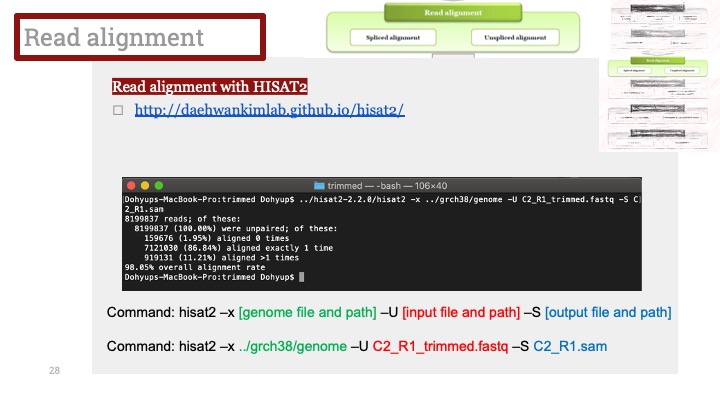
아래는 제가 직접 쓰는 예제 코드입니다.

레퍼런스는 celldex 레퍼런스를 쓰고 있으며, 본인의 데이터 종류에 따라서 여러가지의 레퍼런스를 불러올 수 있다는 것이 장점입니다. Mouse 싱글셀 시퀀싱 데이터를 분석 중인데 가장 간편한 방법이 SingleR이었습니다.
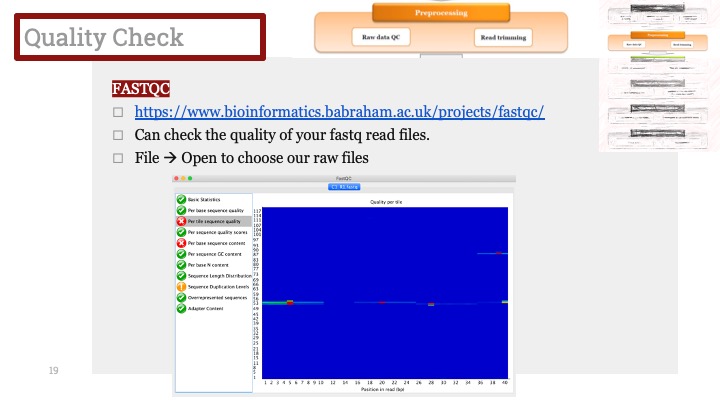
SingleR을 실행시킨 다음, 결과를 heatmap으로 그려볼 수 있습니다.

Heatmap 결과가 위와 같이 나오는 데 이걸 Annotation에 이용할 수 있습니다.
장점은 간편하고 실행 속도가 빠른 편이며 레퍼런스가 다양하다는 것이지만, 단점은 위와 보시다시피 정확한 세포 종류를 못 잡아내는 경우가 있으며, 서로 다른 클러스터가 수치 상으로는 같은 세포 종류가 되는 경우가 있습니다. 그리고 때때로 정말 이상한 결과가 나올 때도 있으니까 수동작업이 항상 필요합니다.
3. Azimuth
Azimuth는 NIH Human Biomolecular Atlas Project의 일환으로 개발된 cell type annotation 도구입니다.
여러가지 레퍼런스를 제공하고 있으며, 가장 큰 장점 중의 하나가 웹에서 구동이 가능합니다. 자신의 데이터를 업로드해서 계산해줍니다. 그만큼 Computing resource가 많이 들기도 하는데, 저는 주로 데이터 용량이 매우 큰 관계로 설치를 하여서 사용하고 있습니다.
장점은 위에서 언급한 것에 추가하여, 실행이 간편하며 특히 Seurat 패키지와 완벽하게 호환됩니다.
단점은 레퍼런스를 여러가지 제공하고 있긴 하지만 웹에서는 제공된 레퍼런스만 사용이 가능하며, 설치버전도 레퍼런스 만들기가 조금 복잡합니다. 또한 계산을 하면 UMAP이 나오는데 이게 좀 안맞을 때가 있습니다. 그래서 수동으로 하나하나 확인해주는 작업이 반드시 필요합니다.

Azimuth를 돌린 결과 UMAP입니다. 아, 그리고 때때로 이게 Seurat에서 작동하다보니 Azimuth를 돌리고 나면 내 원래 데이터를 건드릴 때도 있고, 나는 seurat_clusters에 나온대로 21개의 세포 종류로 annotation하고 싶은데 Azimuth는 resolution이 다를 때가 있습니다 (제가 잘 못하고 있는 거일 수도 있습니다.)
아, 그리고 설치버전은 좀 무거워서 데이터가 좀 큰 경우에 로컬머신에서 돌리기 버거울 수가 있습니다. 저는 주로 HPC 환경에서 돌리고 있습니다.
4. Reference Mapping
이건 딱히 적을 필요가 있나 싶은데 Seurat 패키지 자체에서도 Reference mapping을 제공하고 있습니다.
https://satijalab.org/seurat/articles/multimodal_reference_mapping.html
특히 자주 나오는 조직이나 샘플을 분석 중이라면 이 레퍼런스 맵핑이 유용하게 쓰입니다. 작동하는 방법은 Azimuth와 아주 비슷합니다. 그리고 결과도 Azimuth와 비슷한데, 가끔 Annotation이 다르게 나오는 경우가 있어서 참고용으로 유용하게 쓰고 있습니다.
아래는 제가 직접 쓰는 코드입니다:

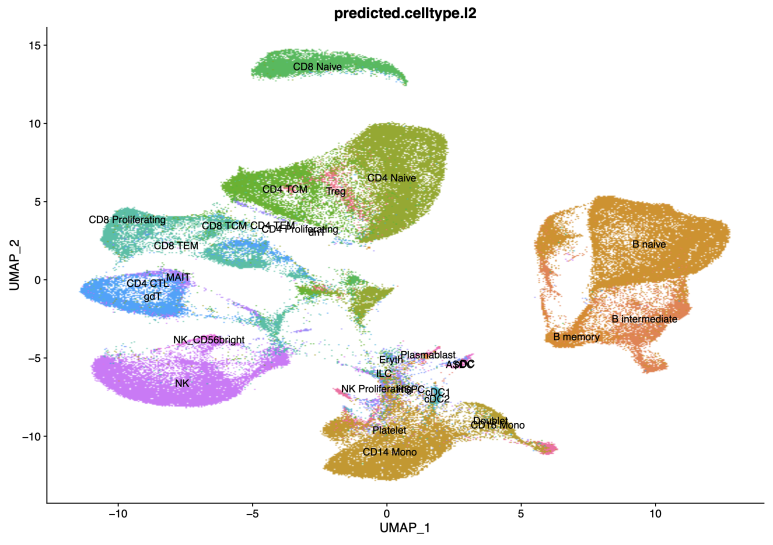
Reference Mapping한 결과물입니다.
결론:
음.... 제가 싱글셀 분석을 하면서 가장 많은 시간을 쏟는 파트이자 가장 어려워하는 파트가 이 Cell type annotation인 것 같습니다. DEG나 clustering 처럼 딱 정답이 나오는 것이 아니고 레퍼런스에 따라 비교 방법에 따라 결과도 상이하게 나오는 경우가 있고 더군다나 마지막에는 결국 내가 결정해야하는 거구나 하고 결정해버리기 때문인 것 같습니다. 처음부터 끝까지 자동으로 해주는 Azimuth나 Reference mapping이 있긴 하지만, 그걸 맹목적으로 믿기에는 결과가 이상하게 나올 때가 많습니다 (위의 UMAP처럼요).
그래서 결국에는 어떤 방법을 쓰더라도 꼭 하나이상을 쓴 다음 그걸 수동으로 확인해서 검증하는 과정이 반드시 필요한 것 같습니다. 그리고 자신이 분석하고 있는 샘플의 특성을 잘 알고 각각 cell type의 특성이 어떤 것인지 잘 이해하고 있으면 더욱 도움이 되는 것 같습니다.
'Biological Science > Bioinformatics' 카테고리의 다른 글
| 싱글셀 Seurat 데이터로 PCA 만들어보기 (feat. PLS-DA) (1) | 2023.05.12 |
|---|---|
| R에서 Seurat 싱글셀 데이터로 GSEA 해보기 (8) | 2023.02.23 |
| 싱글셀 RNASeq 데이터에서 DoubletFinder로 더블렛 없애기 (5) | 2022.11.07 |
| 싱글셀 시퀀싱 을 이용한 RNA Velocity란 (1) | 2022.07.19 |
| Single Cell RNA-Seq Analysis (4) | 2022.07.02 |