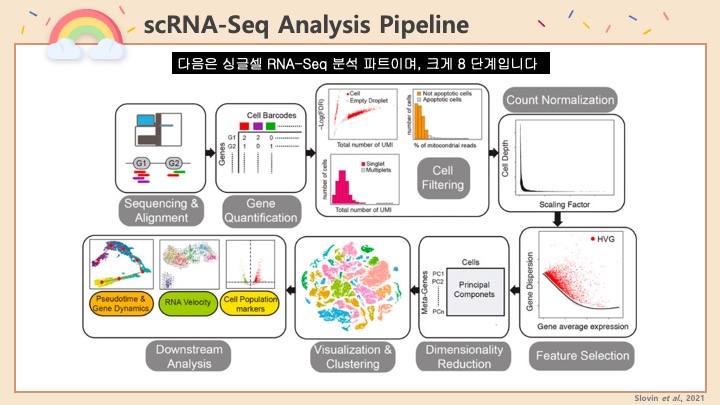
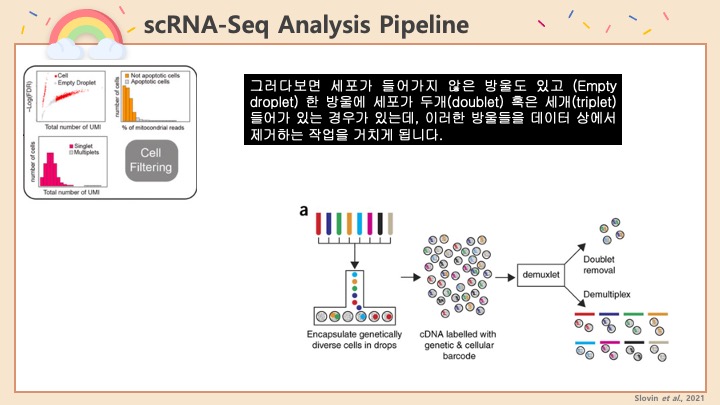
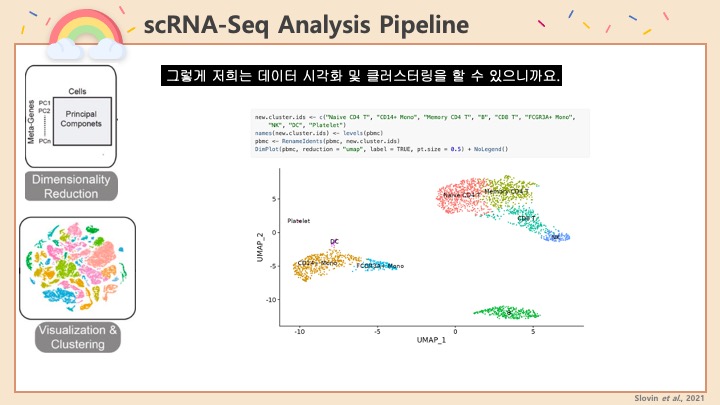
#scRNASeq 분석을 진행하던 도중에 UMAP을 보니 데이터에 #Doublet 이 제법 많이 있다는 사실을 알게 되었습니다. #UMAP 에서 보면 서로 다른 클러스터 사이에 브릿지같은 연결 다리가 보이죠? 그게 서로 다른 두 종류의 세포가 같은 방울 안에 묶여서 같이 시퀀싱 된 더블렛이라고 볼 수 있습니다.

이렇게 데이터 상에서 더블렛이 나타나는 경우에 다음 단계의 분석에 영향을 미칠 수도 있고, 클러스터링이 어려울 수가 있습니다. 그래서 #Seurat 에서는 기본적으로 feature number 혹은 gene number 등에 범위를 줘서 어느정도 doublet을 거르고 있습니다.

하지만 데이터의 성격이나 품질에 따라서 이러한 스크리닝으로도 더블렛이 제거가 안되는 경우가 있습니다. 그럴 때에 따로 Doublet 을 찾아내는 프로그램을 사용하게 됩니다. 이러한 프로그램에는 여러가지가 있지만 저는 그 중에 #DoubletFinder 라는 프로그램을 선택해서 사용하였습니다. 이 소프트웨어를 쓴 이유는:
- 사용 설명이 비교적 자세하고 친절하게 되어있음 (그렇지 않은 프로그램들이 참 많죠 ㅠㅠ)
- 비교적 최근까지 업데이트가 이루어짐
- Seurat 파이프라인과 연계가 간편함 (대부분의 다른 프로그램들은 SingleCellExperiment 스트럭쳐로 옮긴 다음에 실행을 해야해서, 데이터 구조를 변화를 시켜서 실행시킨 다음, 결과가 나온 후에 다시 Seurat 파이프라인으로 옮기는 과정이 들어가야 합니다.)
우리 학교 사람이 만들었어요.
이 소프트웨어는 아래의 링크에서 무료로 구하실 수 있습니다. R 패키지로 설치도 가능하구요.
https://github.com/chris-mcginnis-ucsf/DoubletFinder
GitHub - chris-mcginnis-ucsf/DoubletFinder: R package for detecting doublets in single-cell RNA sequencing data
R package for detecting doublets in single-cell RNA sequencing data - GitHub - chris-mcginnis-ucsf/DoubletFinder: R package for detecting doublets in single-cell RNA sequencing data
github.com
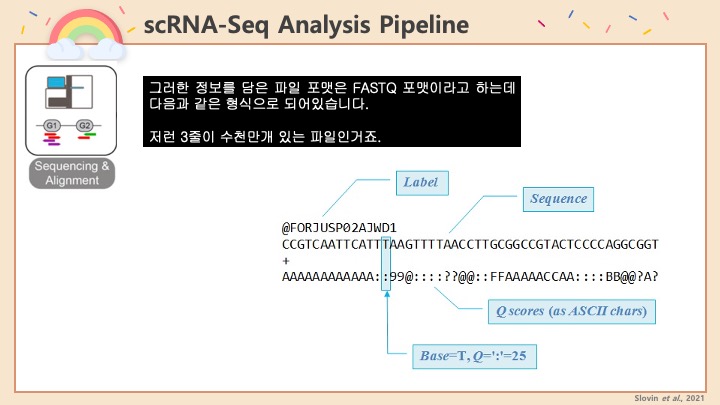
DoubletFinder에 관한 배경이나 설명, 알고리즘은 위의 웹페이지에 자세하게 설명이 되어 있으니 넘어가고 실질적으로 사용하는 과정만 기록하겠습니다.
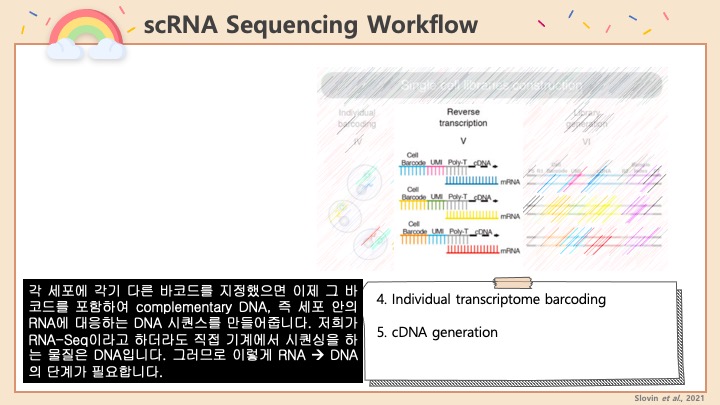
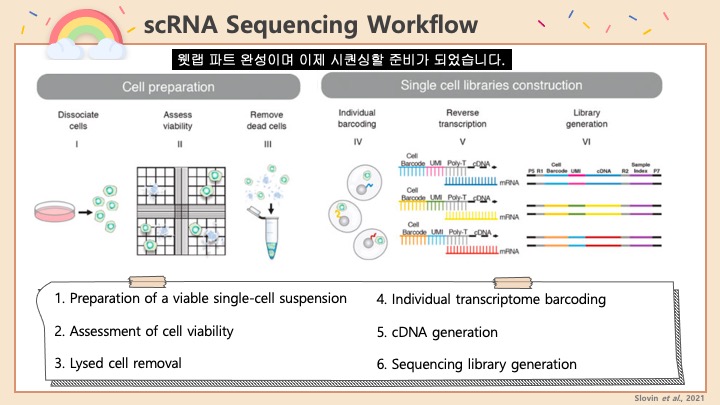
1. 일단 Seurat package를 불러들입니다. 만약 샘플끼리 이미 Integration을 했다면, 더블렛을 제거하고 다시 Integrate를 하는 것을 권장합니다. 아래의 R 코드는 Cellranger에서 count를 마치고난 결과를 읽어들여서 Seurat object로 만드는 것부터 시작합니다.
library(dplyr)
library(patchwork)
library(DoubletFinder)
#메모리를 많이 잡아먹을 수 있기 때문에 사용 메모리를 늘려줍니다
options(future.globals.maxSize = 8000 * 1024^2)
#파일을 불러옵니다
raw.data<-Read10X(data.dir="./p10v3/filtered_feature_bc_matrix/")
#Seurat object를 만듭니다
myData<-CreateSeuratObject(counts=raw.data, project = "myData", min.cells = 3, min.features = 200)
#다음 두줄은 나중에 합쳐서 분석을 위해 정보를 추가했습니다
myData$patient<-"p10"
myData$visit<-"v3"
#익숙한 방법으로 mt 유전자들을 마크하고 걸러냅니다. 이때, Seurat 기본 튜토리얼에서는 5% 이상은 걸러내라고 적혀있는데, 실제 나중에 나온 실험 논문을 찾아보면 이 수치가 종마다, 조직마다 다르니까 찾아보시는 게 좋습니다
myData[["percent.mt"]]<-PercentageFeatureSet(myData, pattern = "^MT-")
VlnPlot(myData,features=c("nFeature_RNA","nCount_RNA","percent.mt"),ncol=3)
myData<-subset(myData,subset=nFeature_RNA > 200 & nFeature_RNA<4500 & percent.mt<15)
2. 그런 다음에 이제 노멀하게 PCA 및 UMAP 등을 돌려서 프로세싱해줍니다.
#기본 Seurat 파이프라인입니다. PCA dimension이나 Cluster resolution 등은 샘플에 맞게 알아서 조절해주세요. 기본으로 쓰셔도 무방합니다.
myData<-NormalizeData(myData)
myData <- FindVariableFeatures(myData, selection.method = "vst", nfeatures = 2000)
all.genes <- row.names(myData)
myData<-ScaleData(myData, features = all.genes)
myData<-RunPCA(myData)
myData<-RunUMAP(myData, dims = 1:30)
myData<-FindNeighbors(myData, reduction = "pca", dims = 1:30)
myData<-FindClusters(myData, resolution = 0.55)
3. 이제 DoubletFinder 홈페이지에 있는 직접 실행시키는 예제가 나오는데요. 변수 이름은 굳이 바꾸지 않았습니다.
sweep.res.list_myData<-paramSweep_v3(myData, PCs = 1:30, sct = FALSE)
sweep.stats_myData<-summarizeSweep(sweep.res.list_myData, GT = FALSE)
bcmvn_myData<-find.pK(sweep.stats_myData)
#이 시점에서 그래프가 나옵니다. pK를 계산해주는 그래프인데, 그래프만으로는 최대값 지점을 한눈에 알기가 어려우므로 다음 줄을 실행시킵니다
bcmvn_myData


4. 이전 단계에서 pK 최대값 지점을 확인한 후에 다음 코드를 실행시킵니다. 여기서 붉은색 숫자는 샘플 안에 Doublet이 얼마나 들어있는지를 예측하는 퍼센트입니다. 이 수치는 시퀀싱 테크닉마다 다르고, 샘플 내의 세포 갯수에 따라 또 다릅니다. 저의 경우에는 10X를 이용했는데 10X Genomics 홈페이지에 다음과 같이 퍼센트가 나와있습니다. 자신 샘플의 세포 갯수에 따라 조절해주세요.
homotypic.prop <- modelHomotypic(myData@meta.data$nFeature_SCT)
nExp_poi<-round(0.085*nrow(myData@meta.data))
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))

5. 이제 실제로 Doublet을 구해봅시다. 아까 찾은 pK를 붉은색 숫자에 알맞게 넣어줍니다. 저희의 경우는 0.005였죠.
myData <- doubletFinder_v3(myData, PCs = 1:30, pN = 0.25, pK = 0.005, nExp = nExp_poi, reuse.pANN = FALSE, sct = FALSE)
#위의 코드를 실행하고 나면, myData의 구조를 다음 코드로 확인해봅니다
head(myData[[]])
#그 후에 아래의 pANN_0.25_0.005_763 을 자신의 데이터에 맞는 이름으로 고쳐주면 됩니다
myData <- doubletFinder_v3(myData, PCs = 1:30, pN = 0.25, pK = 0.005, nExp = nExp_poi.adj, reuse.pANN = "pANN_0.25_0.005_763", sct = FALSE)
head(myData[[]])
#그리고, DimPlot을 만들어 Doublet과 Singlet을 관찰해 봅시다 물론 DF.classifications 뒤에 오는 숫자는 알맞게 고쳐줘야합니다DimPlot(myData, group.by = "DF.classifications_0.25_0.005_763", raster = FALSE)

6. 이제 실제로 Doublet이 몇개인지 Singlet이 몇개인지 전체는 몇개인지 알아봅시다. 이런 수치들은 그때그때 적어서 기록으로 남겨두는 편입니다
length(myData@meta.data$DF.classifications_0.25_0.005_763)
length(myData@meta.data$DF.classifications_0.25_0.005_763[myData@meta.data$DF.classifications_0.25_0.005_763=="Singlet"])
length(myData@meta.data$DF.classifications_0.25_0.005_763[myData@meta.data$DF.classifications_0.25_0.005_763=="Doublet"])
7. 마지막으로 Singlet만 골라서 rds 형식으로 저장합니다.
myData_sub<-subset(myData, subset = DF.classifications_0.25_0.005_763 == "Singlet")
saveRDS(myData_sub, file = "DoubletRemoved/p10v3.rds")
DoubletFinder를 이용하여 더블렛을 제거하고 다시 저장하는 방법을 알아보았습니다.
끝
'Biological Science > Bioinformatics' 카테고리의 다른 글
| R에서 Seurat 싱글셀 데이터로 GSEA 해보기 (8) | 2023.02.23 |
|---|---|
| 싱글셀 시퀀싱 클러스터 Annotation 방법과 팁 (3) | 2023.01.06 |
| 싱글셀 시퀀싱 을 이용한 RNA Velocity란 (1) | 2022.07.19 |
| Single Cell RNA-Seq Analysis (4) | 2022.07.02 |
| 싱글셀 RNA-Seq에서의 Batch Effect (0) | 2022.06.18 |